|
SÍNTESIS:
Se realizó un estudio retrospectivo, descriptivo y longitudinal, dirigido a la búsqueda de todos los pacientes portadores de Carcinoma de Células de Merkel en un período de 13 años (1989-2002). Los objetivos que perseguimos fueron: distribuir nuestra casuística acorde a las características clínicas y patológicas de esta entidad, estadificar nuestra serie según el TNM actual, calcular el valor estadístico y supervivencia de algunas variables pronósticas de interés, determinar la supervivencia global de la casuística incluida en el estudio y relacionar nuestra serie con otras patologías cutáneas y/o viscerales.
Para ello fueron seleccionados 10 casos, cuyo diagnóstico anatomo-patológico presuntivo fue elaborado previamente con técnica de hematoxilina y eosina en el Departamento de Anatomía Patológica del Instituto Nacional de Oncología y Radiobiología (INOR) , siendo el diagnóstico positivo confirmado mediante técnicas de inmunohistoquímica fuera de la institución, aplicándosele a la mayoría de los casos una batería compuesta de CK-20, TTF-1, CK 7, EMA, NSE, Cromogranina A y B, Sinaptofisina, NF-L, CD 99, Proteína S-100, HMB 45 y LCA. Se elaboró una base de datos para el procesamiento de la información. Se utilizaron medidas de resumen para datos cualitativos (porcentajes) y cuantitativos (Media Aritmética, Desviación Estándar).Las funciones de supervivencia fueron estimadas por el Método de Kaplan Meier, y contrastadas con el test de Log Rank. Se determinaron además, las medianas de supervivencia y los intervalos de confianza (95%) correspondientes. Se empleó un nivel de significación de 0.05 en las estadísticas inferenciales. La información se presentó resumida en tablas, figuras y gráficos. Para el procesamiento y análisis de los datos nos auxiliamos del Software Profesional SPSS 11.5. Se utilizó como procesador de texto Microsoft Word.
En nuestra casuística la edad osciló entre 47 y 87 años con una media de 72,10 años, hubo una distribución equitativa de los sexos (50 % masculinos y 50 % femeninos). El color claro de la piel predominó en el 80 % de los casos y un 20 % presentó la piel bronceada. El sitio de predilección de ocurrencia de estos tumores fue la región de la cabeza y el cuello (50%), seguido de las extremidades (30%) y tronco (20%), el tamaño de nuestras lesiones osciló entre 1.1cm y 8 cms con una media de 4.36 cms, todos nuestros pacientes fueron encontrados en etapa I y II del desarrollo de la enfermedad, hubo una distribución casi equitativa según los subtipos histológicos , cobrando mayor número de casos los de células pequeñas e intermedias (40% cada uno). La invasión epidérmica por parte del tumor en forma de ulceración predominó en un 60 % de la casuística sobre aquellas lesiones que no la comprometieron (40 %). Un borde infiltrativo de crecimiento estuvo presente en el 90 % de los casos, la profundidad de invasión tumoral alcanzó la grasa hipodérmica en 6 casos. Del total de nuestra casuística, 8 pacientes fallecieron, 3 de ellos por patologías intercurrentes y 5 por enfermedad incontrolable específica. La supervivencia global en éste estudio mostró un tiempo medio de 5.55 años en un intervalo de confianza del 95 % entre 2.31 - 8.80 y una supervivencia libre de enfermedad de 3.40 años en un intervalo de confianza del 95 % entre 0 .56 - 6.25, en las defunciones por actividad tumoral incontrolable. Otras neoplasias de tipo cutáneo se vieron asociadas con estas lesiones como la Queratosis solar y los Carcinomas basales y epidermoides, de forma sincrónica o en colisión.
Índice..
1. Resumen.
2. Introducción.
3. Objetivos.
4. Material y método.
5. Resultados
6. Discusión
7. Conclusiones
8. Recomendaciones.
9. Bibliografía.
I0. Anexos.
INTRODUCCIÓN:
Fue Friedrich Sigmund Merkel (1845-1919), patólogo alemán, el primero en describir la célula de Merkel normal en 1875(1), como una “célula clara en la unión dermoepidèrmica no queratinocìtica, no dendrítica, en íntima proximidad con fibras nerviosas mielinizadas”; la cual encontró en el hocico de los puercos, así como en gansos y patos; postulando que estas células actuaban como elementos mecano receptores en éstos animales, llamándolas “células táctiles” (1)
En la actualidad se describe a la célula de Merkel como una célula clara, grande, redonda a oval, de citoplasma escaso, con núcleo redondo y cromatina finamente dispersa, localizada en la capa basal epidérmica de la piel, anejos cutáneos y mucosas, aisladamente, pudiéndose confundir con un melanocito o una célula de Langerhans o en grupos paralelos a la superficie constituyendo los llamados corpúsculos de Merkel (2).
Su función exacta sigue permaneciendo desconocida, pero su localización en áreas de percepción sensorial, tales como la punta de los dedos, punta nasal y folículos pilosos en humanos y su íntima asociación con axones nerviosos terminales (disco pilar táctil de Pinkus en piel velluda y terminaciones de Merkel-Ranvier de la piel glabra) encontradas en la nariz , membranas mucosas del labio, paladar, genitales y borde libre palpebral de peces, anfibios, reptiles , aves y mamíferos; sitúan a esta célula como un mecano receptor de adaptación lenta tipo I o receptor táctil primario.(3)(4)(5)(6)(7) (8)(9) (10).
El origen de esta célula permanece sin confirmar pero su génesis en la cresta neural es sostenido por modelos de transplantes embriònicos interespecies en pájaros (11), otros plantean su origen en la célula madre totipotencial epidérmica de la capa basal con capacidad de diferenciación epitelial y neuroendocrina. En realidad, la inmunohistoquímica y la microscopìa electrónica muestran marcadores de ambas líneas celulares ,siendo en la primera, la presencia de inmunoreactividad para la Enolasa Neuronal Específica, Calcitonina , Cromogranina y Sinaptofisina las más representativas en la línea neuroendocrina y la positividad para citoqueratinas (CK) de bajo peso molecular 8,18 y 19, las más expresadas por la extirpe epitelial. Igualmente la ultraestructura revela numerosos gránulos de centro denso limitados por membranas típicos de células neuroendocrinas además de conexiones desmosomales y filamentos de CK de origen epitelial. (2)(3)(6)(9)(12).
Fue Cyril Toker en 1972 quien describe cinco casos de un tumor cutáneo primario con una apariencia indiferenciada, postulando así que la lesión representaba una variante de un carcinoma de glándula sudorípara y debía diferenciarse de las metástasis de una neoplasia visceral. Dado el patrón de crecimiento trabecular del tumor él lo llamó “Carcinoma de Células Trabeculares”. (13). En 1978 Tang y Toker encuentran por microscopìa electrónica gránulos de centro denso típicos de la célula de Merkel y otros tumores de células neuroendocrinas, sugiriendo que estas neoplasias mostraban diferenciación neuroendocrina y por lo tanto sugerían un origen en las células de Merkel de la piel; así lo llamaron Carcinoma Neuroendocrino Primario de Piel o Carcinoma de Células Pequeñas Primario de Piel.(14) En 1980 De Wolf-Peeters sugirió el nombre de Carcinoma de Células de Merkel (CCM) (15) y actualmente las designaciones más aceptadas son la de Carcinoma Neuroendocrino Primario de Piel o Carcinoma de Células de Merkel (16). Otras denominaciones anteriormente usadas fueron las de Carcinoma Indiferenciado Primario de Piel, Carcinoma anaplàsico primario de piel, Tumor Neuroepitelial Primario de Piel, APUDoma Cutáneo de las siglas en inglés (Amine Precursor Uptake Decarboxylation System) y Merkeloma (16).
En la actualidad son pocas las teorías que no dan crédito al origen de esta neoplasia en la célula de Merkel normal, alegando diferencias en la distribución anatómica en cada caso y la localización casi exclusivamente dérmica de los carcinomas comparada con las células de Merkel ubicadas siempre dentro de los epitelios (2).
En Estados Unidos se reporta una incidencia anual de 470 nuevos casos comparadas con 31000 casos anuales de Melanoma Maligno Cutáneo (MMC) (17); constituyendo menos del 1% de todas las neoplasias malignas cutáneas, siendo cien veces menos frecuente que el MMC (18)(19), con una incidencia total ajustada a la edad de 0,44 casos por 100 000 habitantes al año (20) , mostrando la raza blanca una incidencia anual de 0.23 por 100 000 habitantes comparada con el 0.01 por 100 000 habitantes en la raza negra (17). En Cuba no existen reportes estadísticos de esta entidad, aunque en archivos de Patología del Instituto Nacional de Oncología y Radiobiología (INOR) aparecen registrados 14 casos desde el año 1989 hasta el 2002.
La etiología de esta neoformación es desconocida, aunque como en el MMC; ésta se ha visto vinculada a una exposición solar incrementada (21), de ahí que la radiación ultravioleta B juege un papel importante en la producción de la proteína mutante P53 derivada de dicho gen supresor tumoral, producto del daño al DNA, donde ésta puede encontrarse hasta en un 20% de los casos (22). Este estudio está correlacionado clínicamente con su significativa incidencia en la raza blanca, su localización en áreas foto-expuestas en la mayoría de los casos (23), e histopatológicamente por su estrecha vinculación a Carcinomas de Células Escamosas, Carcinomas de Células Basales, Queratosis Actínica, y Enfermedad de Bowen. Otras fuentes de radiación, son atribuidas a los rayos ultravioletas A, en pacientes frecuente afectados con Psoriasis, donde es cien veces más (25), así como en pacientes con quemaduras faciales provocadas por radiación infrarroja durante el tratamiento por Acné juvenil (24) (25). La segunda hipótesis habla de la Célula Madre Totipotencial Epidérmica de la Capa Basal con capacidad de diferenciación dual; estos trabajos se apoyan en la asociación sincrónica en colisión o metasincrónica con los Carcinomas de Células Escamosas y/o Carcinomas de Células Basales antes expresados, además de la frecuencia a mostrar líneas de diferenciación escamosas o basales, en el seno de una misma lesión (26) (27). Otros factores etiológicos se atribuyen a la exposición a sustancias nocivas como el arsénico, viéndose una afiliación con el arsenismo crónico (28), particularmente en áreas geográficas donde el agua está contaminada en altas concentraciones, como una vez ocurrió en la costa sudeste de Taiwán (29). El quinto elemento etiológico guarda estrecha relación con una condición inmunológica incompetente, en la cual estados de inmunosupresión mantenidos como sucede en pacientes con trasplantes de órganos (30) (31) (32) (33), esta entidad constituye el 0,9 % de todas las neoplasias malignas de novo, aparecidas en su seguimiento, mostrando una edad media aproximada de 46 años, siendo esta mucho más baja que la presentada en los casos esporádicos (34) , su relación con el MMC es de 6:1 comparada con la población general de 65:1 (34) y su comportamiento biológico es mucho más agresivo con una mortalidad anual cercana al 50% (35); otras asociaciones son con el SIDA (36) (37) y neoplasias malignas previas que afectan el sistema inmunológico como Leucemias (23) , Mielomas Múltiples (38) y Linfomas no Hodgkin (39) (40) , además el 25 % de los CCM., podrían asociarse posteriormente con otras neoplasias malignas no cutáneas comparado con solo el 5,8% de los casos con MMC (41) . Por último la demostración a veces de remisión espontánea tanto del tumor primario, como de las metástasis a ganglios linfáticos regionales, presumiblemente habla de algún estado de inmunidad alterada. (42) (43)
La lesión elemental primaria que caracteriza clínicamente éstas lesiones viene representada por un nódulo, nódulo exofítico o placa indurada de 2 a 200 mm, con una media entre 7 y 12 mm (44) (45) de color azul-violáceo a rojo púrpura, comparable muchas veces a un forúnculo, pústula, hemangioma o angiosarcoma (46) (47); de superficie brillante, a veces con telangiectasias superficiales remedando un Carcinoma de Células Basales (44) (45) (48). Estas lesiones pueden ser únicas o múltiples, con una presentación sincrónica o metasincrónica, indoloras y su crecimiento puede ser tan rápido como 2 meses o muy lento, con reportes hasta de 2 años de evolución (44) (45). El sangramiento y la ulceración superficial ocurren tardíamente y constituyen signos de enfermedad avanzada (49). Otra presentación frecuente es la de un crecimiento nodular subcutáneo, el cual puede remedar clínicamente un quiste epidérmico de inclusión, ganglión o un histiocitoma fibroso benigno (45) (50). Macroscópicamente estas lesiones muestran una consistencia firme o dura, la superficie de corte revela una coloración gris a blanquecina, a veces con áreas de hemorragia; puede haber extensión descendente del tumor a la hipodermis, fascia y músculo subyacente y sin un tratamiento oncológico específico ellas pueden tener un crecimiento regional destructivo y ulcerativo masivo. (49) (51) (52) De esta forma, dada la similitud morfológica de estas lesiones con carcinomas escamosos o basocelulares, el diagnóstico clínico presuntivo nunca es realizado. Como se ha señalado previamente se encuentra descrita la regresión espontánea completa del tumor primario y hasta la fecha se documentan 7 casos después de una biopsia incisional (42) (53) (54) (55) (56) (57) y 5 más en los que la regresión ocurrió en la recurrencia local o regional (Regresión espontánea completa secundaria (58) (59) (60) (61) (43); esto hace que si bien el 75% de los pacientes se presentan típicamente con una lesión primaria secundaria , los restantes casos se presentarán con una enfermedad regional o distante sin tumor primario conocido. (62) (63).
Desde el punto de vista diagnóstico la confirmación de estas neoformaciones no solo descansa en la presencia de un tumor de células redondas y azules al estudio histopatológico, sino en la correlación de estos resultados con el análisis inmunohistoquìmico, ultraestructural y citogenético.
La Inmunohistoquímica permite hacer el diagnóstico positivo y diferencial con otros Carcinomas de Células Pequeñas, Melanomas y Linfomas. Los CCM expresan marcadores de origen epitelial, marcadores de diferenciación neuroendocrina y marcadores neurales. Entre los primeros se destacan fundamentalmente las CKs de bajo peso molecular 8, 18, 19 y 20, siendo la CK 20 positiva en el 97 % de los casos aunque éste marcador también es reactivo en otros tumores de células pequeñas localizados en la glándula salival (60%), cuello uterino (9 %) y pulmones (0.03%), el patrón de tinción tanto en las células de Merkel normales como en los CCM en acúmulos globulares de situación paranuclear o en glóbulos discretos punteados alrededor del núcleo (64) (65),apareciendo excepcionalmente en carcinomas neuroendocrinos viscerales y coincidiendo con la localización al microscopio electrónico de los filamentos intermedios de queratina (13)(66); su positividad excluye las metástasis cutáneas de un carcinoma de células pequeñas pulmonar. El segundo marcador epitelial de importancia es el Factor 1 de transcripción tiroideo (TTF 1), en uso combinado con la CK20 , el cual es un factor de transcripción nuclear expresado por las células del tiroides, pulmón y la mayoría de los carcinomas epidermoides; así , lo encontramos en el 100 % de los tumores carcinoides atípicos, del 83% al 100% de los carcinomas de células pequeñas bronquiales, el 75% de otros carcinomas neuroendocrinos y 72,5 % de los adenonocarcinomas (67), no siendo expresado por el CCM, lo que permite su diagnóstico diferencial. La inmunotinción negativa para la CK 7 y su presencia en el carcinoma microcítico pulmonar constituye otro elemento diagnóstico diferencial; por último, el Antígeno Epitelial de Membrana puede verse hasta en el 90% de los CCM. (16)(68). Dentro de los marcadores de diferenciación neuroendocrina encontramos positividad de un 50 % a 100 % con la Enolasa Neuronal Específica (NSE) (16)(69), Cromogranina B y A en un 100 % y 72% respectivamente (70), Secrotoneunina en un 22 % y Sinaptofisina en un 39 %, otros neuropéptidos menos constantes y de reactividad focal son la Somatostatina, Calcitonona, ACTH, Sustancia P, Gastrina y Péptido Intestinal Vaso activo (71). Entre los marcadores neurales se describe expresividad de más de un 90 % para la Proteína Neurofilamento L (NF-L) la cual al igual que la CK 20 se dispone en glóbulos paranucleares discretos; otro antígeno neural con reactividad menos constante es el Polipéptido M (16). La positividad en más de un 90% de los tumores Neuroectodérmicos Periféricos Primitivos (PNET) al anticuerpo monoclonal CD 99 permite su distinción del CCM en casos con sospecha de metástasis cutánea (72). La constante inexpresión de los CCM para los marcadores melanocíticos HMB 45 y Proteína S-100, así como el Antígeno Leucocitario Común (LCA), los excluye del Melanoma Maligno de Células Pequeñas y del Linfoma primario o secundario de piel (73)(69). En resumen la detección inmunohistoquímica de CKs de bajo peso molecular y en particular de la CK 20 en compañía de marcadores neuroendocrinos y del antígeno neural Proteína Neurofilamento L hacen posible el diagnóstico positivo de esta entidad , permitiendo su exclusión del Carcinoma De Células Pequeñas de pulmón metastático (CK 7 Y TTF-1 positivos)así como de otros Carcinomas Neuroendocrinos (TTF-1 positivo), PNET (CD 99 positivo),Melanoma Maligno de Células Pequeñas (S-100 y HMB-45 positivos) y Linfomas (LCA positivos) .
Al microscopio electrónico estas neoplasias conservan detalles similares a la célula de Merkel normal, y se describen como redondas a ovales, conteniendo en su citoplasma gránulos neurosecretorios uniformes de 80 a 200 nm, de centro denso y limitados por una membrana; estos gránulos se sitúan hacia la periferia del citoplasma o en cortos procesos citoplasmáticos espinosos de aspecto dendrítico que ellas poseen hacia su extremidad, siendo éste un marcador diagnóstico ultra estructural de tumor neuroendocrino sin excluir la posibilidad de una metástasis cutánea de origen visceral (74). Así el único hallazgo distintivo que lo separa de otras neoplasias neuroendocrinas es la presencia de una malla de agregados globulares de filamentos intermedios de localización paranuclear o yuxtanuclear correspondientes a queratinas y proteínas de neurofilamentos (74) (13) (66). Como otros hallazgos de menor interés se describen la yuxtaposición entre las células tumorales, así como tonofilamentos y uniones intercelulares complejas tipo desmosomales entre ellas y los queratinocitos aledaños. (13) (66).
En el orden citogenético aparecen alteraciones entre un 17% a un 63% de los casos; así la pérdida de la heterozigosidad por translocación y delección del brazo corto del cromosoma 1 en la región 3, banda 6(1p36) se describe hasta en un 40% de las lesiones (75)(76)(77)(78), aunque esta anomalía está implicada también en la patogénesis de tumores de la cresta neural como el Melanoma Maligno, el Neuroblastoma y el Feocromocitoma (75)(79)(80)(81). Otra anomalía frecuente es la pérdida de la heterozigosidad en el brazo corto del cromosoma 3, región 2, banda1 (3p21), la cual está también implicada en más del 90 % de los pacientes con Carcinoma de Células Pequeñas del Pulmón (79) (82). Otras delecciones y ganancias cromosómicas menos estudiadas son: Delección del brazo pequeño del cromosoma 3, delección del brazo largo del cromosoma 5, delección del brazo pequeño del cromosoma 8, delección del brazo largo del cromosoma 10. delección del brazo largo del cromosoma 11, delección del brazo largo del cromosoma 13, delección del brazo pequeño del cromosoma 17, ganancia del brazo largo del cromosoma 1, ganancia del brazo largo del cromosoma 3, ganancia del brazo pequeño del cromosoma 5, ganancia del brazo largo del cromosoma 8 y ganancia del cromosoma X (83) (76).
El tratamiento del CCM se apoya en su sistema de estadiamiento patológico, de manera que para aquellos tumores confinados a la piel menores de 2 cms de diámetro mayor (Etapa I) se debe realizar una excisión local amplia entre 25 y 30 mm de márgenes laterales, por 20 mm de profundidad debido al alto riesgo de recurrencia local cicatrizal (44) (45) (84) (85). En el caso de lesiones de la cara y el cuello donde aparecen involucradas áreas cosméticas y sensitivas algunos autores aplican la cirugía micrográfica de Mohs, la cual permite un control histológico de los márgenes quirúrgicos a través de la biopsia por congelación, minimizando la extensión de la excisión (86). Dada la elevada tendencia a las metástasis a los ganglios linfáticos regionales algunos trabajos recomiendan la disección ganglionar regional profiláctica en aquellos casos localizados en cabeza y cuello con tumores mayores de 1.5 cms de diámetro mayor (23), otros, sólo cuando el tumor contiene 10 o más mitosis por campo de gran aumento, se comprueba la existencia de invasión linfática y/o la lesión tiene histología de células pequeñas (87), Cotlar et, al: la recomiendan en tumores con más de seis semanas de evolución (88). En lesiones de la línea media de la cara ,dado el drenaje linfático bilateral algunos estudios recomiendan la disección radical de cuello bilateral; los resultados del valor terapéutico de ésta técnica son escasos (89)(90)(91); por una parte los patrones anatómicos del drenaje linfático en ésta región son muy ambiguos lo que dificulta la disección del nódulo, pero por otra , estudios preliminares han demostrado que éste procedimiento tiene valor predictivo sobre el estado anatómico del resto de los ganglios de la cuenca regional y si estos resultados logran ser confirmados a gran escala, como con el MMC, esta técnica proporcionará una alternativa menos mórbida de tratamiento y un estadiamiento patológico más preciso en los pacientes con enfermedad regional oculta (92)(93)(94). Dada la radiosensibilidad del tumor en ésta etapa, la tercera modalidad terapéutica lo constituye la radioterapia adyuvante, así algunos trabajos recomiendan la irradiación post-excisión del sitio primario y de los ganglios regionales (95) (96), otros sólo la indican cuando las lesiones tienen más de 15 cms de diámetro mayor, los márgenes quirúrgicos están por debajo de 2 mm, o el tumor muestra evidencias de invasión linfática (97). En lesiones confinadas a la piel pero mayores de 2 cms de diámetro mayor (Etapa II) se recomienda en primer lugar la excisión de la lesión primaria con disección de los ganglios linfáticos regionales, en segundo lugar la radioterapia adyuvante del área post-excisión del sitio primario y de la región ganglionar, y en tercer lugar se considera la quimioterapia aunque los resultados con ésta última arma son poco satisfactorios (98) (99). Para la Etapa III y IV donde la enfermedad ha alcanzado los ganglios linfáticos regionales o hay toma visceral a distancia, la radioterapia y los agentes quimioterapéuticos juegan sólo un papel paliativo (98) (99).
Considerando toda la temática expuesta, llena de una profunda atracción científica, entre otros aspectos, por su infrecuencia epidemiológica y su poca divulgación, decidimos elaborar un estudio dirigido a describir las características clínicas y patológicas de estas lesiones en nuestra serie, agruparla según el sistema de estadiamiento patológico TNM vigente, determinar la sobrevida global, y según algunas variables pronósticas de interés, relacionar los casos con otras patologías cutáneas y/o viscerales, y caracterizar las defunciones por el curso irreversible de la enfermedad.
Objetivos:
1) Distribuir nuestra casuística acorde a las características clínicas y patológicas de esta entidad.
2) Estadificar nuestra serie según el TNM actual.
3) Calcular el valor estadístico y supervivencia de algunas variables pronósticas de interés.
4) Determinar la supervivencia global de la casuística incluida en el estudio.
5) Relacionar nuestra serie con otras patologías cutáneas y/o viscerales.
Material y método:
Se realizó un estudio retrospectivo, descriptivo y longitudinal dirigido a la búsqueda de todos los pacientes portadores de CCM en un período de 13 años (1989-2002), seleccionándose 10 casos, cuyo diagnóstico anatomo-patológico presuntivo fue elaborado previamente con técnica de hematoxilina y eosina en el Departamento de Anatomía Patológica del Instituto Nacional de Oncología y Radiobiología (INOR) , siendo el diagnóstico positivo confirmado mediante técnicas de inmunohistoquímica fuera de la institución. Para estos fines, los bloques de parafina más representativos fueron enviados al doctor Carlos Monteagudo del Departamento de Patología de la Facultad de Medicina, en Avenida Blanco Ibáñez 17, 46010, Valencia, España y a la doctora Anna Wankowicz-Kalinska del Departamento de Patología del centro médico Académico de la Universidad de Amsterdam , Meibergoreef 9, 1105 AZ, Holanda; donde en ambos centros simultáneamente se realizó técnica de Avidina-Biotina-Inmunoperosidasa, para el desenmascaramiento antigénico se aplicó técnica de autoclave Heating a 1.5 atmósferas durante 10 minutos en buffers citrato a PH 6. No disponemos de las fuentes comerciales y diluciones de los anticuerpos monoclonales empleados, pero en la mayoría de los casos se aplicó una batería compuesta de CK-20, TTF-1, CK 7, EMA, NSE, Cromogranina A y B, Sinaptofisina, NF-L, CD 99, Proteína S-100, HMB 45 y LCA. Las láminas de Inmunohistoquímica no están tampoco disponibles.
Para evaluar la invasión linfático-vascular del tumor en aquellos casos donde éste parámetro era dudoso, empleamos técnica de histoquímica convencional con la coloración de Verhoff para las fibras elásticas de las paredes linfovasculares.
1- ) Localización:
Aplicamos la misma distribución topográfica de Medina-Franco H, et al: (19) dividiéndose de la siguiente manera:
Cabeza - Cuello.
Tronco.
Extremidades.
2- ) Tamaño:
Consiste en la mensuración del tumor en centímetros; basándonos en el sistema de estadificación patológico actualmente empleado para estas lesiones. (100).Lo dividimos en:
< 2cms.
>2cms.
3- ) Etapa Patológica:
Utilizamos el sistema de cuatro estadios patológicos propuestos por Allen PJ, et al: (100) adaptado a los sistemas habituales de estadificación del American Joint Committe on Cancer.
Estadio I: Confinado a la piel, tumor < 2cm. de diámetro máximo. (Bajo riesgo)
Estadio II: Confinado a la piel, tumor ≥ 2cm. de diámetro máximo. (Bajo riesgo)
Estadio III: Diseminación ganglionar. (Alto riesgo)
Estadio IV: Metástasis a distancia. (Alto riesgo)
4- ) Subtipo Histológico:
Células pequeñas:
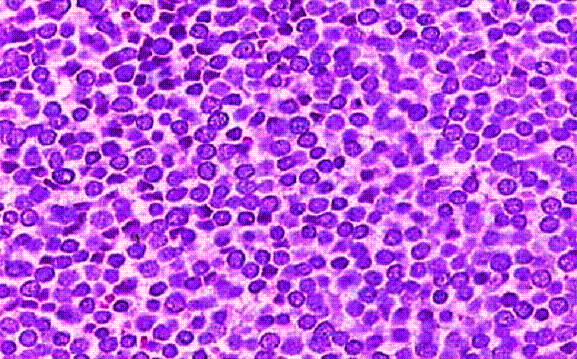
Su aspecto es semejante a un tumor de células redondas pequeñas y azules de localización visceral (101) (52) (102) mostrando un patrón de crecimiento en sábanas difusas (102)(67), se destaca el artefacto de aplastamiento nuclear dado por incrustaciones basofílicas alrededor de los vasos sanguíneos intratumorales (efecto de Azzopardi).(102)(67)(95)(103) Las células se disponen de manera compacta con moldeamiento nuclear; sus núcleos basofílicos son redondos u ovales, regulares de 2 a 3 veces más grandes que el de los linfocitos maduros, con un patrón de cromatina nuclear granular y fino, con apariencia de polvo dispuesto hacia la periferia dándole a los núcleos una tonalidad pálida difusa (núcleo vesicular).El citoplasma es escaso, anfófilo o basófilo, de bordes mal definidos . (51)(16)(104)(105)
Células intermedias:
Se distingue del primer subtipo en que el citoplasma es algo más abundante que el de una célula pequeña, aunque menos que el del subtipo trabecular (51) (102) (67). A diferencia del subtipo anterior hay tendencia de la lesión a rodear y no destruir los anejos cutáneos especialmente los folículos pilosos (52) (102) (104), además de que las células se disponen de manera menos compacta por lo que el moldeamiento nuclear no es patente. (95)(103)(105)
Trabecular:
Se destaca por su patrón de crecimiento en trabéculas finas interconectadas, separadas por un estroma fibrovascular delicado, el cual cobra magnitud con el uso de coloraciones especiales (Retículo y Van Geason). (13)(52)(102)(104). Este patrón puede verse secundariamente en la periferia de los subtipos anteriores de manera inconspicua (101) y hay tendencia a la disposición perianexial como en el anterior subtipo (52) (51) (16).Participan células pequeñas e intermedias, pero también hay formas poligonales y fusiformes de citoplasma mas abundante y bien definido con núcleo grande, ovalado y nucleolo prominente, las cuáles pueden agruparse formando rosetas de Homer Wright. (52)(102)(67)(95)(103).
Combinado:
Son aquellos tumores conformados por células de diferentes tamaños, adoptando diversos patrones de crecimiento entremezclados, lo que dificulta clasificarlos en un tipo histológico específico. (101) (51) (16) (52) (102) (67) (95) (103) (104) (105)
5- ) Compromiso Epidérmico:
Se refiere al modo de invasión del tumor en dicha estructura y la respuesta de la misma. Se consideró en:
1) Ausente: Ausencia de células tumorales.
2) Presente como:
a) Hiperplasia Reactiva: Acantosis irregular, papilomatosis e hiperqueratosis subyacente a una neoplasia del dermis (68) (74)
b) Ulceración: Pérdida de la continuidad epidérmica de la piel producto de la invasión de una tumoración dérmica (68) (74)
c) Patrón Pagetoide: Infiltración de células neoformativas en forma individual y/o en acúmulos a distintos niveles de dicho estrato, respetando muchas veces la integridad de la capa basal, pero en íntima relación con un tumor dérmico. (106)
d) Epidermotropismo: Infiltración de células tumorales aisladas rodeadas por un halo claro o en grupos a distintos niveles de dicho estrato, con poca o ninguna espongiosis, procedente de una tumoración dérmica.
e) Intraepitelial: Proliferación de células neoplásicas dispuestas en islotes sueltos, confinadas en la epidermis sin la existencia de ninguna neoplasia subyacente (107) (108) (109)
6- ) Nivel de Invasión:
Se refiere al crecimiento vertical en profundidad según los planos histológicos afectados. Este parámetro se dividió según Mott R T, et al: en: (104)
Dermis Reticular
Hipodermis
Fascia
Músculo Subyacente
7- ) Número de Mitosis:
Se identificó el área de mayor actividad mitótica y posteriormente se realizó el conteo del número de mitosis en 1 mm² con una lente de 100 x 40 dioptrías, siendo 5 campos de gran aumento el equivalente de 1 mm². Este parámetro se evaluó en: (13) (38) (74 (110) (111) (112)
<10m / CGA
>10m / CGA
8- ) Infiltrado Linfoplasmocitario:
Este indicador se refiere a la infiltración de elementos linfo-plasmocitarios tanto dentro como alrededor del tumor. Se operacionalizó de la siguiente forma según Kossard S, et al: (101) (113)
Intenso: infiltrado inflamatorio dentro y alrededor de la lesión
Ligero: infiltrado inflamatorio en el perímetro de la lesión
Nulo: infiltrado inflamatorio ausente.
9- ) Bordes: Se refiere al modo de crecimiento en la interfase tumor – tejido conservado. (67) (102) (51)
Infiltrativos: Son aquellos que se ven a menor aumento como márgenes deshilachados , que corresponden a mayor aumento con células tumorales aisladas y sueltas diseminadas fuera de la masa tumoral principal, como cordones estrechos percolando las fibras colágenas del dermis.
Circunscritos: Son aquellos que crecen de manera pujante o expansiva comprimiendo las fibras colágenas del dermis adyacente con poca o ninguna evidencia de diseminación radial de células tumorales sueltas.
10- ) Invasión Linfo-vascular:
Es la invasión de células tumorales dentro de la microvasculatura cercana al tumor por colisión con el endotelio (invasión incipiente) o ruptura del mismo y llenado de la luz (invasión patente). (110) (114) (115)
Presente
Ausente
Análisis estadístico:
Se elaboró una base de datos para el procesamiento de la información. Se utilizaron medidas de resumen para datos cualitativos (porcentajes) y cuantitativos (Media Aritmética, Desviación Estándar).
Las funciones de supervivencia fueron estimadas por el Método de Kaplan Meier, y contrastadas con el test de Log Rank. Se determinaron además, las medianas de supervivencia y los intervalos de confianza (95%) correspondientes. Se empleó un nivel de significación de 0.05 en las estadísticas inferenciales.
La información se presenta resumida en tablas, figuras y gráficos
Para el procesamiento y análisis de los datos nos auxiliamos del Software Profesional SPSS 11.5. Se utilizó como procesador de texto Microsoft Word.
RESULTADOS:
En el período estudiado, 14 pacientes fueron diagnosticados con un CCM y 10 casos cumplieron con los criterios de inclusión de nuestra investigación.
La edad osciló entre 47 y 87 años con una media de 72,10 años; 7 casos fueron mayores de 70 años (70 %). La edad mayor de 70 años se relacionó en un 60 % del total de los casos con bordes de crecimiento tumoral Infiltrativos, así como defunciones por curso irreversible de la enfermedad. Esta variable no mostró ningún valor estadístico respecto a la supervivencia.
Se encontró una distribución equitativa en el orden de los sexos, con 5 hombres (50 %) y 5 mujeres (50 %), lo que hizo que no existiera ninguna vinculación con alguna variable en específico, no obstante el 80 % de los hombres mostraron mayor tendencia a tener lesiones superiores a 2 cms. Tampoco este parámetro tuvo significancia estadística.
El color de la piel se vio reflejado en 8 pacientes con piel blanca (80 %) y 2 bronceadas (20 %). El color blanco se relacionó con bordes Infiltrativos de crecimiento (90%), invasión linfático-vascular (80%), fallecidos por curso fatal (80%), y en el 60 % de las veces con lesiones superiores a 2 cms, ulceradas, con un nivel hipodérmico de profundidad de invasión y más de 10 M/CGA, además de aparecer en todos los casos con satelitosis, metástasis en tránsito y a ganglios linfáticos regionales (100 %). Al realizar el estudio estadístico este indicador no aportó ninguna significación.
El sitio de predilección de ocurrencia de estos tumores fue la región de la cabeza y el cuello (50%), seguido de las extremidades (30%) y tronco (20%), no existiendo ninguna asociación con otras variables en especial, no obstante la localización aunque no mostró valor de significación estadística (p = 0. 8228), las lesiones situadas en el tronco mostraron una sobrevida más baja que las ubicadas en las extremidades , con una media de 2.84 años (IC 1.12 – 4.57) y 6.24 años (IC 1.42 -11.06) respectivamente.
El tamaño de las lesiones osciló entre 1.1cm y 8 cms con una media de 4.36 cms, el 60 % fueron mayores de 2 cms (6 pacientes) y el 40% menor de 2 cms (4 pacientes). Las lesiones mayores de 2 cms se relacionaron en un 80 % con fallecidos por curso fatal y un 60 % a pacientes de piel blanca, con lesiones de bordes infiltrativos, con más de 10 M/CGA e invasión linfático-vascular. Al realizar el análisis de supervivencia, encontramos que el punto de corte de peor pronóstico en el estrato de dimensiones tumorales fue a partir de los 3 cms, con una significancia estadística de p = 0.0462, (Ver tabla 1), situándose en cuarto lugar, precedido por el estado anatómico de los ganglios linfáticos regionales, la etapa patológica y la presencia de respuesta inflamatoria hospedero - tumoral, con una sobrevida media de 1.72 años IC(1.35 - 2.09) para lesiones mayores de 3 cms, comparada con 7.65 años IC(3.75 - 11.54) en neoplasias menores de 3 cms y un tiempo medio de recaída de 0.93 años IC(0.50 - 1.36) y 6.24 años IC(1.39 - 11.09) en cada caso. Ver gráficos 1 y 2.
Según el sistema de estadificación TNM, todos nuestros casos al momento diagnóstico fueron encontrados en etapa I y II del desarrollo de la enfermedad, lo que hizo que éste parámetro mostrara los mismos resultados y asociación de variables que el tamaño tumoral, no obstante, considerando el tamaño tumoral en 2 cms como el punto de separación entre una etapa y otra, obtuvimos una sobrevida en 5 años de un 75 % y 33.3 % en cada estadio sucesivo y al realizar el cálculo estadístico en 12 años de seguimiento, éste se ubicó en el segundo lugar luego del estado anatómico de los ganglios linfáticos regionales con un alto valor de significación (p =0.0147), (Ver tabla 1), y una diferencia en la sobrevida de 8.96 años IC (5.22 - 12.69) para la etapa I y de 1.67 años IC (1.28 - 2.06) para la etapa II, con un tiempo de recaída de 6.24 años IC(1.39 - 11.09) y 0.93 años IC(0.50 - 1.36) sucesivamente. Ver gráficos 3 y 4.
Hubo histopatológicamente una distribución casi equitativa según los subtipos histológicos cobrando mayor número de casos los de células pequeñas e intermedias (40% cada uno); esto hizo que este indicador tampoco mostrara ninguna vinculación de importancia con los otros parámetros analizados. Desde el punto de vista estadístico no encontramos valores de significación estadística.
La invasión epidérmica por parte del tumor en forma de ulceración predominó en un 60 % de la casuística sobre aquellas lesiones que no la comprometieron (40 %), mostrándose solamente vinculación en un 80 % con los fallecidos por actividad tumoral irreversible. Tampoco este parámetro mostró significancia estadística.
El borde infiltrativo de crecimiento estuvo presente en el 90 % de los casos (9 pacientes) viéndose reflejado en el 100 % de las defunciones por irreversibilidad del proceso, 80 % de los pacientes con piel de color blanca, con invasión linfático-vascular y el total de fallecidos; así como en el 60 % de pacientes mayores de 70 años con lesiones superiores a 2 cms y más de 10 M/CGA, además el 100 % de las metástasis a ganglios linfáticos regionales se relacionaron con esta variable; sin embargo, este parámetro no mostró significación estadística.
La profundidad de invasión tumoral alcanzó la grasa hipodérmica en 6 casos, existiendo un 100 % de aproximación con la enfermedad loco-regional metastásica y en un 80 % con las metástasis a distancia y muertes por curso irreversible de la enfermedad; no obstante, aunque este factor pronóstico no tuvo valor estadístico (p = 0.1181), aquellas neoplasias que invadían hasta la grasa hipodérmica tuvieron una sobrevida media más baja que el resto de los otros niveles de invasión encontrados : 2.83 años (IC: 0.82 - 4.83) vs 4.05 años (IC:0.28 – 1.95).
Un índice mitótico mayor de 10 mitosis por campo de gran aumento se vio reflejado en 7 pacientes de nuestra casuística, apareciendo asociado con el 60 % de los casos de piel blanca y lesiones superiores a 2 cms, con bordes infiltrativos de crecimiento, invasión linfático-vascular y defunciones por múltiples causas; además el 100 % de los pacientes masculinos se relacionó con este índice. De igual manera este indicador tampoco mostró significancia estadística.
No se reflejaron diferencias distintivas en la distribución de la casuística según las 3 categorías de respuesta linfoplasmocitaria presentes en estas neoformaciones pero se destacó la ausencia de infiltrado inflamatorio en el 100 % de los casos con toma de los ganglios linfáticos regionales y metástasis en tránsito, en el 80 % de las metástasis a distancia y defunciones por actividad tumoral irreversible, así como el 60 % de las satelitosis. Paralelamente obtuvimos una significancia de gran valor (p = 0.0272), (Ver tabla 1), quedando en tercer lugar, precedida del estado anatómico de los ganglios linfáticos regionales y la etapa patológica, donde lesiones con infiltración linfoplasmocitaria presente reflejaron un tiempo de sobrevida medio de 9.23 años IC(5.85 - 12.61) con relación a aquellas neoplasias sin infiltrado inflamatorio (1.52 años IC : 1.11 - 1.94), y un tiempo de recaída ( p = 0.1000 ) de 5.38 años IC(1.07 - 9.68) y 0.86 años IC (0.28 -1.44) en cada caso. Ver gráficos 5 y 6.
La presencia de invasión linfático vascular se vio reflejado en 8 casos (80 %), apareciendo en el 100 % de los fallecidos por actividad tumoral incontrolable; así como en los pacientes con enfermedad loco-regional, el 80% de los casos con piel de color claro y tumores de bordes de crecimiento infiltrativo, así como en el 70% de los fallecidos por causas generales y en el 60% de lesiones mayores de 2 cms con más de 10 M/CGA. Desde el punto de vista estadístico esta variable no aportó ningún efecto diferencial en la supervivencia.
A 4 pacientes se les realizó una excisión quirúrgica incompleta siendo reintervenidos quirúrgicamente en un período entre 20 y 45 días; la recurrencia cicatrizal ocurrió en 4 casos entre 8 y 10 meses, 3 fueron únicas y en 1 paciente, ésta sucedió en 2 ocasiones separadas por un intervalo de 2 meses cada una. Los satélites microscópicos estuvieron contenidos en la biopsia inicial en 4 casos, además otro paciente presentó 1 lesión semejante 12 meses después de la excisión. Estos tipos de lesiones se relacionaron en el 100 % de los casos con pacientes de piel clara y neoplasias que invadían hasta la grasa hipodérmica, de bordes infiltrados, con respuesta inflamatoria nula, invasión linfático-vascular, además de vincularse con la generalidad de los fallecidos; también todos los casos con satelitosis tuvieron tumores ulcerados.
Tanto la recurrencia local cicatrizal como los satélites macros y microscópicos no aportaron valor estadístico de importancia.
Las metástasis en tránsito se reflejaron en 3 casos, 2 de ellas fueron únicas y en 1 paciente éstas se vieron de manera múltiple y metasincrónicas, con un intervalo de aparición entre 6 y 22 meses después del diagnóstico inicial en todos los casos. Este parámetro mostró la misma asociación de variables que la recurrencia local cicatrizal y la satelitosis, aunque el estudio estadístico de éste factor no arrojó un valor de significación (p=0.1691) encontramos diferencias en los valores de sobrevida medios en aquellos casos con metástasis en tránsito (1.67 años, IC: 1.24 - 2.10), de los que no la presentaron (7.87 años, IC: 4.15 - 11.59).
Las metástasis a los ganglios linfáticos regionales sucedieron en 4 casos, con exéresis y radioterapia en 3 pacientes y 1 tratado solamente con radioterapia y quimioterapia; siendo el intervalo de aparición entre 3 y 24 meses. Este indicador pronóstico mostró la misma asociación de variables que la recurrencia local cicatrizal, la satelitosis y las metástasis en tránsito con excepción de la ulceración. Estadísticamente esta variable ocupó el primer lugar con una significancia de p = 0.0101, (Ver tabla 1), con una sobrevida media en casos con enfermedad regional negativa de 7.66 años IC:(3.77- 11.55) y de 1.34 años IC:(1.07-1.60) para pacientes con ganglios linfáticos regionales positivos, así como una significancia de p = 0.0459 en la sobrevida libre de enfermedad con un tiempo medio de recaída de 4.71 años IC:(0.95 - 8.47) en casos con enfermedad regional negativa y 0.60 años IC:(0 .21- 1.00) para pacientes con ganglios linfáticos positivos. Ver gráficos 7 y 8.
Las metástasis a distancia se vieron reflejadas en 5 pacientes en un tiempo comprendido entre 10 y 25 meses después del diagnóstico inicial, siendo el pulmón y el hígado los órganos más afectados. Asombrosamente este parámetro no mostró valor estadístico.
Del total de nuestra casuística, 8 pacientes fallecieron, 3 de ellos por patologías intercurrentes y 5 por enfermedad incontrolable específica, en estos últimos el intervalo de tiempo transcurrido entre el diagnóstico de la enfermedad y el evento fatal fue entre 14 y 30 meses, estando el 100 % de estos casos asociadas con enfermedad visceral sistémica.
La supervivencia global en éste estudio mostró un tiempo medio de 5.55 años en un intervalo de confianza del 95 % entre 2.31 - 8.80 y una supervivencia libre de enfermedad de 3.40 años en un intervalo de confianza del 95 % entre 0 .56 - 6.25, en las defunciones por actividad tumoral incontrolable. Ver gráficos 9 y 10.
Otras neoplasias de tipo cutáneo se vieron asociadas con éstas lesiones, destacándose en 2 casos con biopsias anteriores, la existencia de Queratosis solar, otro paciente tuvo además 2 carcinomas basales y epidermoides aparecidos posteriormente y en 1 caso se vio la presencia sincrónica y en colisión con el CCM, un carcinoma epidermoide bien diferenciado grado I.
Discusión:
Se registran casos de CCM con un rango etario desde 7 hasta 104 años (116) (117), aunque éste aparece básicamente en personas de edad avanzada con un rango etario que oscila entre la séptima y octava décadas de la vida y una media de 69 años (19) (17) (118) (119) (89) (120) (121), solamente el 5 % de los pacientes tienen menos de 50 años y dentro de ellos se destacan los sujetos trasplantados, donde la edad de aparición es mucho más joven, con una media al diagnóstico de aproximadamente 10 años menor que la de los pacientes no inmunocomprometidos (34) (122) ; así Penn I, et al: (34) en una serie de 41 casos tuvo una media etaria al diagnóstico de 53 años y el 29 % de los pacientes no superaban los 50 años (122). Nosotros tuvimos un 70 % de pacientes mayores de 70 años con una media de 72,10 años y el resto de las series consultadas integradas por pacientes no inmunosuprimidos, coinciden en tener una media etaria mayor de 60 años (19) (17) (118) (119) (89) (120) (121), también coincidimos en una casuística analizada, en la cual la edad más joven fue de 47 años como el menor de nuestros pacientes (39). Algunos estudios bioestadísticos arrojan resultados controvertidos al señalar la edad como variable pronóstica (23) (44) (120) (123) (124); Shaw y Rumball (84) en 1991 revisaron 30 publicaciones con información suficiente para propósitos pronósticos en 234 casos y llegaron a la conclusión que las edades menores de 50 años cursan con desfavorables destinos ; otro estudio analizado de 34 pacientes mostró similares resultados (125), sin embargo la mayor parte de las publicaciones coinciden en señalar un empeoramiento del pronóstico por encima de los 55 a 60 años. (23) (136) (123) (84) (119) (52) (95)
En la mayoría de las grandes series la incidencia de estas neoformaciones es algo mayor en varones, variando el radio de proporciones de 1.4 a 1.5:1 (19) (39) hasta 2.3 a 2.1:1.Esta última relación es tomada del registro de vigilancia epidemiológica de Estados Unidos (17); en una revisión de 875 pacientes se encontró un radio masculino / femenino de 1.5:1 (39), sin embargo , algunos autores han observado mayor incidencia en las mujeres como Pasella J, et al: con 20 casos que mostró una relación hombre - mujer de 0.4 : 0.6 (127), Savage P, et al: con 22 casos, esta fue de 0.2:0.7 (126), y Bishof M, et al: con 17 casos, de 0.4:0.5 (128).Otras series no encuentran diferencias (146) como resultó en nuestra casuística. En algunos estudios de riesgo, el sexo masculino conlleva un pronóstico desfavorable (84) y Pitale M, et al: (19) en una serie de 309 pacientes concluye que el sexo masculino constituye una variable pronóstica independiente adversa con una marcada tendencia a las recurrencias locales y metástasis distantes aunque no tuvo diferencias en las recurrencias regionales; Shaw JH, et al: (84) y Meeuwissen JA, et al: (124) coinciden en éste señalamiento, aunque otras series no pueden confirmarlo (52) (103) (26) (123). Para nosotros este parámetro no tuvo significación estadística, sin embargo el sexo masculino estuvo presente en el 80 % de las lesiones mayores de 2 cms de diámetro mayor, este último un factor de mal pronóstico.
No existe ninguna contradicción en señalar a los pacientes de piel blanca como los de mayor incidencia en la enfermedad como expresamos en la introducción; (21) de ahí que esto, más su estrecha aparición en áreas fotoexpuestas (23) y con lesiones por daño actínico, (51) (85) (129) formen parte integral de la evidencia etiológica que juega la radiación solar en estos casos; aunque está descrita su rara aparición en personas de piel oscura, ésta se origina principalmente en áreas no expuestas a la luz solar como son el tronco, extremidades acrales y región genital (21). Dada la alta incidencia de estos tumores en la piel clara esta cualidad no ofrece asociación estadística con otras tonalidades de piel, razón por la cual no figure en ningún estudio pronóstico.
Estas lesiones aparecen casi siempre en zonas expuestas al sol y en una revisión de 1034 casos (19) su orden de frecuencia fue: cabeza y cuello (47 %), extremidades (33 %), tronco (10 %), genitales y región perianal (2 %), apareciendo en menos del 1% en otras localidades. Dentro del área de cabeza y cuello siguen el siguiente orden: región periorbitaria (46 %), mejilla (29 %), párpado (18 %), frente (17 %), labio (9 %), orejas (7 %), nariz y cuello (5 %), así como cuero cabelludo (4 %) (146). Las extremidades inferiores sobrepasan a las superiores en un 22 % y 16 % cada una (146) y raramente se documentan apariciones en áreas no expuestas al daño solar como son la cavidad nasal , mucosa oral de las encías y paladar duro, piel retroauricular, mucosa de la vulva y del pene.(146) (107) (130) (131). Nosotros coincidimos en tener el 50 % de los casos en la región de cabeza y cuello. Como variable pronóstica algunos trabajos señalan que los tumores del área del tronco son los que llevan la peor parte, particularmente los situados en la región vulvar y perianal , estos últimos por su mayor accesibilidad a los canales vasculares y linfáticos (119) (130) (132), otros describen un elevado número de recidivas locales cicatrizales , satelitosis y metástasis en tránsito en CCM de extremidades inferiores ; (52) (74) y en sitios como cabeza y cuello O'Connor, et al: (133) lo atribuyen principalmente a su relación con márgenes quirúrgicos incompletos en cirugías realizadas que fueron más cosméticas que exeréticas , aunque en la casuística de 234 casos analizada por Shaw JH, et al: éste fue el sitio más desfavorable. (84). Esta variable no tuvo relevancia en nuestro estudio estadístico como en otros análisis (23) (26) (123) (124); no obstante, como en los trabajos de Ott MJ, et al: (119), Tai PT, et al: (130) y Poulsen M, et al: (132), las lesiones situadas en el tronco (tórax anterior y posterior) mostraron un peor pronóstico que las ubicadas en las extremidades , con una sobrevida media de 2.84 años (IC:1.12 - 4.57) y 6.24 años (IC:1.42 - 11.06) respectivamente.
El CCM suele presentarse habitualmente como una lesión menor de 2 cms, aunque su tamaño promedio está entre 0.7 y 1.2 cms (49) (51) (52); en la serie de Skelton HG, et al: (26) de 132 casos ésta osciló entre 0.4 a ,18 cms; Medina-Franco H, et al: (19) en una revisión de 1024 pacientes encuentra diámetros entre 0.2 a 20 cms, y se han descrito dimensiones mayores hasta de 23 cms (134). Según la frecuencia en las dimensiones del tumor, Akhtar S, et al: (39) reporta un 50 % de pacientes ≥ 2 cms de diámetro mayor y Sibley RK, et al: (114) encuentra un 70 % de tumores de iguales características.
La mayor parte de los trabajos publicados están de acuerdo en señalar el punto de corte de peor pronóstico en el estrato de dimensiones tumorales a partir de los 2 cms (41) (67) (26) (95) (135); sin embargo, otros lo encuentran después de 3 cms (23) (101) y aún otros después de los 5 cms (104) (125), por otra parte Ott MJ, et al: (119) y O'Brien PC, et al: (136) no hallaron significación estadística en el tamaño tumoral de sus series. En nuestro análisis la dimensión tumoral de peso pronóstico en el futuro de los pacientes fue a partir de los 3 cms con una significancia de p =0.0462, situándose en cuarto lugar, precedido por el estado anatómico de los ganglios linfáticos regionales, la etapa patológica y la presencia de respuesta inflamatoria hospedero - tumoral, con una sobrevida media de 1.72 años para lesiones mayores de 3 cms, comparada con 7.65 años en neoplasias menores de 3 cms y un tiempo medio de recaída de 0.93 años y 6.24 años en cada caso coincidiendo con los estudios realizados por Pitale M, et al: (23) y Llombart B , et al: (101)
No existe acuerdo general sobre el estadiamiento más apropiado que debe ser aplicado en estas lesiones, pero tomando en cuenta el orden de progresión de la enfermedad similar muchas veces al del MMC, en 1991 Yiengpruksawan A, et al: (44) propuso un modelo basado en 3 etapas, el estadío I tomaba en cuenta el tamaño tumoral de 2 cms como el punto de corte en el estrato de dimensiones para subdividir a este en 2 grupos, el I a serían lesiones < 2cms, y el I b ≥ 2 cms, una vez que la enfermedad alcanzaba los ganglios linfáticos regionales era un estadio II, y si estaba presente en los ganglios linfáticos distantes y/o habían metástasis viscerales, se estadiaba como una etapa III; con esta estadificación la sobrevida en 2 años era del 67% (estadío I a), 59% (estadío I b) , 49% (estadío II) y 23% (estadío III) respectivamente. (95) (130) Más recientemente en el año 2005, Allen PJ, et al: (100) propone una modificación al sistema, el cual encajaría mejor con el TNM actualmente aplicado a las neoplasias cutáneas no melanocíticas y melanocíticas según el American Joint Committe on Cancer, esta se centra básicamente en separar los 2 subgrupos dimensionales en estadios independientes, así la etapa I toma en cuenta solo lesiones < 2 cms, la etapa II aquellas ≥ a 2 cms, la etapa III a las metástasis presentes en los ganglios linfáticos regionales, y la etapa IV a las metástasis a los ganglios linfáticos distantes y/o metástasis viscerales; con esta estadificación la sobrevida en 5 años es del 81% (estadío I), 67% (estadío II), 52% (estadío III), y 11% (estadío IV) en cada uno. Este autor señala que para lograr los mejores resultados de supervivencia es necesario el empleo de la técnica del ganglio centinela en los casos de enfermedad localizada (etapa I y II) a fin de obtener información del estado anatómico de los ganglios linfáticos regionales y la potencial posibilidad de micro-metástasis ocultas, de esa manera se logra una estadificación patológica más exacta en el progreso de la enfermedad.
Según análisis realizados en grandes series el 70 % al 80 % de los pacientes se encuentran en estadios I y II (tumores < ó ≥ 2 cms) al momento diagnóstico. (39) (95) (130), nosotros no tuvimos casos en etapas III y IV y la sobrevida en 5 años fue más baja que la descrita por Allen PJ, et al: (100) para los estadios I y II con un 75 % y 33.3 % respectivamente. Al realizarse el cálculo estadístico en 12 años de seguimiento, la estadificación obtuvo un alto valor de significación (p = 0.0147) ubicándose en el segundo lugar luego del estado anatómico de los ganglios linfáticos regionales, con una diferencia en la sobrevida de 8.96 años para la etapa I y de 1.67 años para la etapa II, con un tiempo de recaída de 6.24 años y 0.93 años sucesivamente. Estos resultados coinciden con las observaciones realizadas por muchos autores quienes la consideran como el único factor pronóstico predictivo o el mas importante relacionado con la sobrevida libre de enfermedad y con la sobrevida global al momento diagnóstico (19) (39) (85) (137) (138).
A menor aumento pueden destacarse en estas neoplasias grandes lóbulos de células tumorales que experimentan necrosis central con o sin microcalcificaciones, destacándose las pseudorosetas perivasculares a mayor aumento; son los subtipos de células pequeñas e intermedias los que contienen mayor actividad mitótica, oscilando ésta entre 3 y 15 mitosis por mm², sus células se disponen de manera compacta con moldeamiento nuclear, mostrando éstos últimos uno o más nucleolos pequeños o inconspicuos (51) (16) (52) (67) (95) (102) (103). Nosotros no vimos el efecto de Azzopardi, los núcleos vesiculares, las rosetas ni las calcificaciones, elementos presentes en las 4 variantes de células pequeñas diagnosticadas, como tampoco la disposición perianexial del tumor en las 4 variantes de células intermedias. Algunos estudios describen ésta última variedad como la más frecuente, representando el 50 % de las casuísticas (67) (102) (104) (105).Además que ambos subtipos cobran en algunas series el comportamiento clínico más agresivo, no obstante, Smith DE, et al: (139) con un estudio de 35 casos y Akhtar S, et al: (39) con otro de 10 pacientes encuentran una mayor incidencia en la variedad de células pequeñas con el 65 % y 60 % respectivamente. Nosotros tuvimos una distribución casi homogénea entre los subtipos de células pequeñas, intermedias y combinado, que hizo que ninguno superara el 40 % de la totalidad de las lesiones, paralelamente esta misma distribución restó significación al evaluar los eventos fatales. Como predictor pronóstico algunos estudios consideran la variedad de células pequeñas (23) (123) (136) (26) (84) como un factor pronóstico independiente de sobrevida global el cual además si es conjugado con los bordes infiltrativos de crecimiento potencializa un 10 % más su acción predictiva; para otros sus resultados son controvertidos ya que la mayor parte de las veces éstos tumores exhiben combinaciones de 2 o 3 subtipos que los hacen difíciles de clasificar en busca de una significación pronóstica (101)(105); Fernández-Figueras MT, et al: (140); ha informado por otra parte, que la asociación de neoplasias de células intermedias con un alto índice mitótico está directamente asociado a las metástasis loco-regionales y distantes, según sus resultados. Como valor pronóstico esta variable tampoco aportó ningún valor de significación estadística.
Aunque la mayoría de las veces los CCM aparecen en la dermis y/o el tejido celular subcutáneo, la invasión epidérmica puede ser observada en menos de un 10 % de los casos y una vez invadida puede adoptar diferentes patrones como es principalmente la ulceración o más raramente la hiperplasia epidérmica reactiva (68) (74); otros cuadros más raros son el Epidermotropismo donde Walsh NM ,et al: (141), lo describe presente hasta en el 34 % de su casuística simulando una Micosis Fungoides, el patrón pagetoide simulando un MMC in situ, subtipo extensivo superficial, una enfermedad de Paget extramamaria o una Enfermedad de Bowen (106), así como una rara forma intraepidérmica remedando un Epitelioma Intraepitelial de Borst- Jadassohn.(108) (109). El compromiso epidérmico por parte del tumor no constituye una variable de riesgo tanto independiente como subordinada a otras de reconocida significación estadística en modelos pronósticos univariables y multivariables, según pudimos apreciar al menos en la bibliografía consultada, diferencia ésta, significativamente, de los MMC donde la ulceración constituye el segundo determinante de la lesión primaria del sistema TNM después del Índice de Breslow y el único factor de riesgo tumoral que modifica el pronóstico en la enfermedad regional. Nosotros tuvimos un ligero predominio de lesiones ulceradas (60 %) sobre aquellas que no alcanzaban la epidermis (40 %), contrario a lo reportado, no existiendo ninguna asociación importante entre la ulceración y otras variables clínico-patológicas, aunque el 80 % de los fallecidos por curso fatal mostraron lesiones ulceradas. Tampoco este parámetro mostró importancia estadística.
En la mayor parte de la veces los bordes laterales del tumor adoptan un patrón de crecimiento infiltrativo, siendo más evidente en las variables histológicas de células pequeñas e intermedias y aunque más raro también se describen bordes circunscritos de crecimiento representados mayormente en la forma diferenciada o trabecular (51) (52) (67) (95) (102) (103) (104) (105). En la serie de Skelton HG, et al: (26) el 90 % de las lesiones contenían bordes infiltrativos de crecimiento y en otra serie de 34 pacientes (125) este alcanzó el 95 %. Para Tennvall J, et al: (147) que evaluó 17 casos, los bordes infiltrativos de crecimiento se vincularon con una mayor incidencia de neoplasias que invadían en profundidad hasta la grasa hipodérmica, con un alto índice mitótico e invasión linfático-vascular así como con el total de casos con enfermedad regional, a distancia y defunciones por desarrollo irreversible de la enfermedad. La literatura informa que el patrón difuso o infiltrativo de crecimiento cobra valor pronóstico sólo cuando éste se asocia con la variedad histológica de células pequeñas, reduciendo la sobrevida global en un 10 % más que cada una de estas variables analizadas separadamente (23) (123) (84) (136). Esta asociación no cobró ninguna importancia en nuestros resultados, al existir una distribución casi equitativa entre este patrón y todos los subtipos histológicos, además de no aportar tampoco ningún valor estadístico como variable independiente.
Estas lesiones se localizan inicialmente en la dermis, pero en casos avanzados ellos cobran profundidad invadiendo la grasa hipodérmica, fascia y el músculo subyacente percolando entre los fascículos musculares sin destruirlos. (67) (102) Ott MJ, et al: (119) en una casuística de 93 casos encuentra que el 80 % de los pacientes se diagnosticaron en la grasa hipodérmica, Krasagakis K, et al: (142) y Akhtar S, et al: (39) cada uno con una casuística similar a la nuestra encuentran la misma localización en el tejido subcutáneo con el 90 % y 80 % respectivamente.
Para Mott RT, et al: (104) una profundidad de invasión a la hipodermis entraña un pronóstico desfavorable cuando se asocia a una respuesta linfoplasmocitaria nula, como expresamos anteriormente. Por otra parte Koljonen V, et al: (125) afirma que un mayor nivel de invasión tumoral, a partir de la hipodermis se relaciona directamente con las metástasis ganglionares regionales al demostrar que el 100 % de sus casos con enfermedad regional mostraban invasión a la grasa, fascia o músculo esquelético. Nosotros tuvimos un 100 % de aproximación con la enfermedad regional metastásica y un 80 % con las metástasis a distancia y muertes por curso irreversible de la enfermedad en aquellos casos que invadían la grasa hipodérmica, no obstante aunque este factor pronóstico no tuvo valor estadístico (p = 0.1181), aquellas neoplasias con estas características obtuvieron una sobrevida media más baja que el resto de los otros niveles de invasión encontrados (2.83 años).
Suele existir un elevado índice mitótico que oscila entre 3 y 15 M/CGA en estas lesiones; Smith DE, et al: (139) y Pitale M, et al: (23) reportan un índice mitótico mayor de 10 mm² entre un 75 % y 80 % cada uno en sus lesiones, siendo los subtipos histológicos de células pequeñas e intermedias los más representativos (13) (38) (74) (110) (111) (112); nosotros tuvimos el mayor número de casos con > 10 M/CGA en la variedad de células intermedias, aunque con una ventaja mínima del resto de los subtipos histológicos. En otros resultados de pequeñas series (143) este parámetro también cobra mayor significación en la variedad de células pequeñas (80 %). Para Savage P, et al: (126) que evaluó 22 casos, un índice mitótico > 10 M/CGA se relacionó en mayor frecuencia con pacientes mayores de 70 años, del sexo masculino, portadores de lesiones con bordes infiltrativos de crecimiento y respuesta hospedero-tumoral ausente. Skelton HG, et al: (26) en una casuística de 132 casos describe al rango mitótico > de 10 M/CGA como un factor estadísticamente significativo de mal pronóstico, con un efecto independiente en su análisis de regresión multivariable y tumores con < 10 M/CGA, mostraron una sobrevida de 79 % en 5 años y 38 % en lesiones de > 10 M/CGA concluyendo que le rango mitótico tiene una correlación inversamente proporcional con la sobrevida, lo cual lo hace un parámetro estadístico independiente de supervivencia global. Otros trabajos no le dan significación estadística (23) (84) (123) (124). Finalmente otros consideran que esta variable debe ser sustituida por parámetros más precisos como es la expresión en más de un 50 % de marcadores de proliferación celular, como es el KI 67 desarrollado a través de técnicas de inmunohistoquímica , lo que implicaría un pronóstico desfavorable. (101) (140). En nuestro trabajo la relación que este indicador tuvo con otras variables adversas como fueron el gran tamaño tumoral, los bordes infiltrativos de crecimiento, la invasión linfático-vascular y el número de defunciones por curso fatal, en lesiones con > de 10 M/CGA, este parámetro en una casuística más numerosa llegaría a constituir una variable con valor predictivo.
En una serie de 93 casos (141) los CCM con un infiltrado inflamatorio nulo se relacionaron básicamente, con pacientes del sexo masculino que mostraron lesiones superiores a 2 cms, localizadas en el tronco, pertenecientes al subtipo histológico de células pequeñas, con > 10 M/CGA e invasión linfático-vascular.
Un infiltrado inflamatorio nulo se asocia para algunos autores con un pronóstico desfavorable adquiriendo un elevado valor predictivo (86 %), una sensibilidad del 100 % y una especificidad del 63 % cuando éste se conjuga con lesiones que alcanzan la grasa hipodérmica (104). El infiltrado inflamatorio intenso podría ser indicativo de una buena respuesta inmune y por tanto estar ligado a una evolución más favorable como sucede con el MMC (101), no obstante, para otros el grado de infiltración linfoplasmocitaria no necesariamente se relaciona con la intensidad de la respuesta inmune al existir tumores de mal pronóstico con respuesta linfoplasmocitaria activa, lo que hace necesario el uso de marcadores monoclonales de activación linfocitaria para poder medir su capacidad funcional (125). De forma opuesta Skelton HG, et al: (26), mostró las cifras de sobrevidas más bajas en los tumores con infiltrados linfoplasmocíticos intensos y finalmente en otros trabajos este indicador no cobró ningún valor predictivo (52) (105) (119). Nosotros mostramos una significancia de gran valor (p=0.0272) y en lesiones con ausencia de infiltrado inflamatorio linfoplasmocitario obtuvimos las cifras de sobrevida más bajas (1.52 años vs 9.23 años) para un tiempo medio de recaída de 0.86 años y 5.38 años en ausencia o presencia de respuesta hospedero-tumoral respectivamente, ocupando esta variable el tercer lugar, luego del estado anatómico de los ganglios linfáticos regionales y la etapa patológica.
//
La invasión linfática y/o vascular es una característica muy frecuente de estas neoformaciones no existiendo una variedad histológica donde ésta predomine más aunque algunos la señalan frecuentemente en el subtipo de células pequeñas (115), no obstante es un detalle histológico habitual en lesiones avanzadas en tamaño y profundidad (13) (38) (74) (110) (111) (112) (114) (115), encontrando trabajos donde la misma oscila desde un 75 % (141) hasta un 92 % (139). En la serie de Pacella J, et al: (127) de 20 casos esta variable se relacionó mayormente con los casos por encima de 70 años, la enfermedad regional y distante así como el total de fallecidos por curso fatal.
No existe un acuerdo unánime sobre este indicador en pequeñas series (39) (49) (144), aunque en grandes series, su presencia es inversamente proporcional a la sobrevida, incrementa significativamente el riesgo de la recaída local, el compromiso de los ganglios linfáticos regionales, las metástasis distantes y muerte. (26) (101) (125)
La invasión linfático-vascular está asociada con un rango de sobrevida global en 5 años de 25%, comparada con un 50% cuando ésta no existe (125). A pesar de ser ésta un predictor de importancia pronóstica en otras series estadísticamente esta variable no aportó ningún valor en este trabajo.
Shaw JH, et al: (84), considera que la enfermedad loco-regional en forma de metástasis en tránsito y/o a ganglios linfáticos regionales, constituyen variables pronósticas desfavorables y en su estudio de sobrevida para 10 años, ésta fue de un 40 % cuando la enfermedad comprometía solo 1 linfonodo, 26 % cuando ésta oscilaba, entre 2 y 4 ganglios linfáticos regionales y 15 % cuando habían 5 ó más metástasis. En un estudio de 80 casos (124) las metástasis en tránsito y las macrometástasis a linfonodos regionales > 3 cms figuraron variables pronósticas adversas; Ott MJ, et al: (119): considera la recaída loco-regional un indicador de pobre pronóstico; Pitale M, et al: (23) en su estudio de 309 pacientes considera que tanto el satélite, la recurrencia local cicatrizal, las metástasis en tránsito como a ganglios linfáticos regionales no influyeron en la desfavorable evolución de la enfermedad; similarmente otros trabajos no consideran la recaída loco-regional un indicador de mal pronóstico (23) (44) (112) (123). Por otra parte muchos estudios consideran que la presencia de afectación ganglionar es el factor predictivo más importante de las metástasis a distancia y de la supervivencia global, constituyendo el único indicador pronóstico de la enfermedad regional (etapa III) (19) (39) (85) (137) (138); Morrinson WH, et al: (145) documentó una sobrevida media en pacientes sin metástasis ganglionar de 13 meses comparada con 4 meses para aquellos que si la tenían. Este autor concluye que la supervivencia a los 5 años disminuye según aumenta el número de ganglios linfáticos regionales afectados de un 66 % con un ganglio positivo a un 30 % cuando hay más de 4 linfonodos comprometidos; Allen P J, et al: (100) reporta que la supervivencia a los 5 años en pacientes con ganglios linfáticos regionales negativos fue del 97 % frente a un 52 % en aquellos casos con metástasis regional.
Estadísticamente nosotros no encontramos diferencias de sobrevida al evaluar la recurrencia local cicatrizal y la satelitosis como predictores de importancia pronóstica, sin embargo la metástasis a ganglios linfáticos regionales ocupó el primer lugar en valor predictivo (p = 0.0101), con una sobrevida media global de 1.34 años frente a aquellos casos negativos de metástasis ganglionar regional (7.66 años), y un tiempo medio de recaída de 0.60 años vs 4.71 años.
Inicialmente se pensó que el CCM era una lesión de buen pronóstico cuando sólo 1 de los 5 casos de Toker y solo 3 de los primeros 24 casos reportados en la literatura fallecieron (115), sin embargo en la actualidad se considera éste un tumor altamente agresivo y letal con un curso clínico comparable a un MMC de espesor intermedio o grueso (119); a pesar de haber tenido un 30 % del total de fallecidos por actividad tumoral irreversible (50 %) con excisión quirúrgica inicial incompleta, podemos añadir que la supervivencia libre de enfermedad fue de 3.4 años con una sobrevida global de 5.5 años, comportándose esta entidad de manera similar como en otras partes del mundo y esta afirmación quedó apoyada en los resultados de Eftekhari F, et al: (141) que describió una sobrevida media de 4.9 años, Smith DE, et al: (139) de 5.9 años y un tanto parecido , alrededor de 5 años otros autores. (26)(125)
Esta entidad se asocia a una elevada incidencia de otras neoplasias cutáneas y extracutáneas; esta interrelación de lesiones ha sido documentada en los casos de Savage P, et al: (126), Huang HP, et al: (29) y Brenner B, et al: (41) quienes describen una sumatoria de un 40 % y 50 % cada uno entre Queratosis solares y/o Carcinomas basales y/o Carcinomas epidermoides aparecidos antes , durante (sincrónico y/o en colisión) o después del diagnóstico de un CCM; excluyendo de sus resultados otras malignidades viscerales menos frecuentes como fueron Adenocarcinomas de colon, próstata, mama, ovario y procesos linfoproliferativos, nuestra serie se relacionó en un 40 % de los pacientes con las mismas patologías cutáneas y formas de presentación descritas arriba, todas ellas aparecidas en áreas fotoexpuestas y como detalle curioso valdría agregar que en 1 caso apareció de forma integral formando parte macroscópica del mismo tumor un carcinoma de células escamosas, lo cual apoya la tesis sostenida por Hashimoto K, et al: (27) de una célula basal intraepidérmica totipotencial con capacidad de diferenciación dual, la cual pudiera dar origen a este fenómeno, además de la exposición crónica a la radiación ultravioleta B. (51) (85) (141)
Conclusiones:
1) En la caracterización del CCM, la edad mayor de 70 años, el color claro de la piel, la localización en el área de cabeza y cuello y un tamaño tumoral superior a 2 cms. de diámetro fueron variables clínicas dominantes que estuvieron en concordancia con las series consultadas, no coincidiendo la distribución según el sexo, donde las grandes casuísticas señalan una mayor incidencia en varones. Dentro de las variables patológicas inherentes a la neoformación primaria encontramos cifras mas elevadas de lesiones ulceradas que lo habitual, por otra parte un nivel de profundidad de invasión a la grasa hipodérmica, lesiones de bordes infiltrativos de crecimiento, un índice mitótico mayor de 10 M/CGA y la presencia de invasión linfático-vascular fueron características propias de estas neoplasias que la semejaron al resto de los trabajos estudiados.
2) Según el Sistema de estadificación patológica (TNM) aceptado para estas lesiones, el 70 % u 80 % de los pacientes deben estar en una etapa localizada del desarrollo de la enfermedad al momento del diagnóstico (Etapa I y II), según análisis de grandes series, sin embargo, dado lo reducido de nuestra casuística el 100 % de los pacientes se encontraban en estadío I y II al realizarse el diagnóstico positivo y finalmente, a los 5 años de seguimiento encontramos sobrevidas más bajas que las descritas para estas etapas.
3) A pesar de constituir una serie muy corta, en orden decreciente de importancia estadística, las metástasis a los ganglios linfáticos regionales, la estadificación patológica de la enfermedad, la respuesta hospedero-tumoral y el tamaño de la lesión, fueron indicadores pronósticos con valor predictivo en nuestro estudio, lo cual está respaldado en mayor o menor medida con los resultados obtenidos en otros análisis. Aunque la profundidad de invasión tumoral, las metástasis en tránsito y el sitio anatómico de origen del tumor no mostraron valor de significación estadística, se vieron marcadas diferencias en las curvas de sobrevida global al ser dicotomizadas cada una de estas variables, lo cual nos obligó a discutirlas con los resultados obtenidos en otras series donde éstas cobraron valor predictivo. Por otra parte otros factores pronósticos de importancia estadística en muchos trabajos como son la edad, sexo, subtipo histológico de CCM, índice mitótico, bordes de crecimiento del tumor, satelitosis y metástasis a distancia, no cobraron ningún valor en nuestro análisis.
4) Aunque el 30 % de los fallecidos por enfermedad específica tuvo una excisión quirúrgica inicial incompleta, el comportamiento biológico de estas neoplasias resultó ser el mismo que el descrito en la mayor parte de pequeñas y grandes series, es decir alrededor de 5 años luego del diagnóstico patológico de la enfermedad.
5) Un 40 % de nuestra casuística aparecida sobre áreas fotoexpuestas se relacionó con otras neoplasias cutáneas de origen epidérmico como fueron la Queratosis Solar (20 %), el Carcinoma de células escamosas (20 %) y el Carcinoma de células basales (10 %). En uno de los casos el carcinoma epidermoide se presentó de forma sincrónica y en colisión con el CCM, características muy frecuentes encontradas en los reportes de muchas series, y que ponen al relieve el posible papel protagónico de la radiación solar, como uno de los factores etiológicos en la génesis del CCM.
Recomendaciones:
Para lograr una mayor exigencia en la búsqueda de parámetros histopatológicos de importancia pronóstica en pacientes portadores de Carcinoma de Células de Merkel, es de importancia vital el manejo de estos especímenes por un personal técnico-profesional especializado en este tipo de patología, a fin de lograr la mayor calidad en los informes de biopsias, que permitan predecir un pronóstico más efectivo en la sobrevida global, si tomamos en cuenta la significación estadística de algunas variables estudiadas, como lo documentan muchos de los trabajos analizados, y el comportamiento biológico altamente agresivo y letal de estas neoplasias.
Bibliografía:
1. Merkel F: Tastzellen und Tastkörperchen bei den Haustieren und beim Menschen. Archiv für Mikroskopische Anatomie und Entwicklungsmechanik 11:636-652, 1875
2. DK.Kim,K.A.Halbrook. The appearance,density,and distribution of Merkel cells in human embryonic and fetal skin: Their relation to sweat gland and heir follicle development.The Journal of Investigative Dermatology,1995;104:411- 416
3. Winkelmann RK, Breathnach AS. The Merkel cell. J Invest Dermatol. 1973; 60:2-15
4. Camisa C, Wiessmann A. Friedrich Sigmund Merkel, II: the cell. Am J Dermatopathol. 1982; 4:527-535
5. Knox SJ, Kapp DS. Hyperthermia and radiation theraphy in the treatment of recurrent Merkel cell tumors. Cancer. 1988;62:1479-1486
6. Hartschuh W, Weihe E, Yanaihara N, et al. Immunohistochemical localization of vasoactive intestinal polypeptide (VIP) in Merkel cells of various mammals: evidence for a neuromodulator function of the Merkel cell. J Invest Dermatol. 1983; 8: 361-364
7. Smith KR Jr. The Haarscheibe. J Invest Dermatol. 1977;69:68-74
8. English KB. Morphogenesis of haarscheiben in rats. J Invest Dermatol. 1977;69:58-67
9. Munger BL, Halata Z. The sensorineural apparatus of the human eyelid. Am J Anat. 1984; 170:181-204
10. Pinkus F: Über einen bisher unbekannten Nebenapparat am Haarsystem des Menschen: Haarscheiben. Dermatologische Zeitschrift 9:465-469, 1902
11. Grim M, Halata Z: Developmental origin of avian Merkel cells. Anat Embryol (Berl) 202:401-410, 2000
12. Scott SA, Cooper E, Diamond J. Merkel cells as targets of the mechanosensory nerves in salamander skin. Proc R Soc Lond B Biol Sci. 1981;211:455-470
13. Toker C. Trabecular carcinoma of the skin.Arch Dermatol. 1972; 105:107-110
14. Tang CK, Toker C: Trabecular carcinoma of the skin: An ultrastructural study.Cancer 42:2311-2321, 1978
15. De Wolf-Peeters C, Marien K, Mebis J, et al: A cutaneous APUDoma or Merkel cell tumor? A morphologically recognizable tumor with a biological and histological malignant aspect in contrast with its clinical behavoir. Cancer 46:1810-1816, 1980
16. Haag ML, Glass LF, Fenske NA. Merkell cell carcinoma: diagnosis and treatment. Dermatol Surg. 1995; 21:669-683
17. Miller RW, Rabkin CS: Merkel cell carcinoma and melanoma: Etiological similarities and differences. Cancer Epidemiol Biomarkers Prev 8:153-158, 1999
18. Chuang TY, Su WP, Muller SA. Incidence of cutaneous T cell lymphoma and other rare skin cancers in a defined population. J Am Acad Dermatol 1990; 23: 254-256
19. Medina-Franco H, Urist MM, Fiveash J, Heslin MJ, Bland KI, Beenken SW. Multimodality treatment of Merkel cell carcinoma: case series and literature review of 1024 cases. Ann Surg Oncol 2001; 8: 204-208
20. Hodgson NC. Merkel cell carcinoma: Changing incidence trends. J Surg Oncol 2005; 89: 1-4
21. Miller RW, Rabkin CS. Merkel cell carcinoma and melanoma: etiological similarities and diferences. Cancer Epidemiol Biomarkers Prev 1999; 8: 153-158
22. Van Gele M, Kaghad M, Leonard JH, Van Roy N, Naeyaert JM, Geerts ML, Van Belle S, Cocquyt V, Bridge J, Sciot R, De Wolf-Peeters C, De Paepe A, Caput D, Speleman F. Mutation analysis of P73 and TP53 in Merkel cell carcinoma. Br J Cancer 2000; 82: 823-826
23. Pitale M, Sessions RB, Husain S. An analysis of prognostic factors in cutaneous neuroendocrine carcinoma. Laryngoscope 1992; 102: 244-249
24. Lardy F, Gautier C, Etesse-Pichon S, Martin JC, Demeaux H, Geniaux M, et al. Post-radiotherapy cutaneous neuro-endocrine carcinoma. Ann Dermatol Venereol1996;123:464-7
25. Lunder EJ, Stern RS. Merkel-cell carcinomas in patients treated with methoxsalen and ultraviolet A radiation. N Engl J Med 1998 22;339:1247-8
26. Skelton HG, Smith KJ, Hitchcock CL, McCarthy WF, Lupton GP, Graham JH. Merkel cell carcinoma: analysis of clinical, histologic, and immunohistologic features of 132 cases with relation to survival. J Am Acad Dermatol 1997;37:734-9
27. Hashimoto K, Lee MW, D'Annunzio DR, Balle MR, Narisawa Y. Pagetoid Merkel cell carcinoma: epidermal origin of the tumor. J Cutan Pathol 1998;25:572-9
28. Lien HC, Tsai TF, Lee YY, Hsiao CH. Merkel cell carcinoma and chronic arsenicism. J Am Acad Dermatol 1999; 41: 641-643
29. Huang HP, Huang AH, Lee JY. Merkel cell carcinoma: report of three cases.J Formos Med Assoc 1991;90:900-3
30. Urbatsch A, Sams WM Jr, Urist MM, Sturdivant R. Merkel cell carcinoma occurring in renal transplant patients. J Am Acad Dermatol 1999; 41:289-91
31. Plunkett TA, Haris AJ, Ogg CS, McDonald DM, Harper PG. The treatment of Merkel cell carcinoma and its association with immunosuppression. Br J Dermatol1998;139:345-6
32. Formica M, Basolo B, Funaro L, Mazzucco G, Segoloni GP, Piccoli G. Merkel cell carcinoma in renal transplant recipient. Nephron 1994;68:399
33. Stempfle HU, Mudra H, Angermann CE, Weiss M, Reichart B, Theisen K. Rapid growth of cutaneous neuroendocrine Merkel cell carcinoma during treatment of refractory cardiac allograft rejection with OKT3 monoclonal antibody. J Heart Lung Transplant 1993; 12:501-3. abstract
34. Penn I, First MR: Merkel´s cell carcinoma in organ recipients: Report of 41 cases. Transplantation 68:1717, 1999
35. Emilio Suárez Martín. Cánceres Cutáneos en pacientes trasplantados. Dermatología Venezolana 2001; 39(2):213-216
36. An KP, Ratner D. Merkel cell carcinoma in the setting of HIV infection. J Am Acad Dermatol 2001; 45: 309-312
37. Engels EA, Frisch M, Goedert JJ, Biggar RJ, Miller RW. Merkel cell carcinoma and HIV infection. Lancet 2002; 359: 497-498
38. Silva EG, Mackay B, Goepfert H, Burgess MA, Fields RS. Endocrine carcinoma of the skin (Merkel cell carcinoma). Pathol Annu 1984; 19: 1-30
39. Akhtar S, Oza KK, Wright J. Merkel cell carcinoma: report of 10 cases and review of the literature. J Am Acad Dermatol. 2000; 43: 755-767
40. Valdés F, Sánchez-Aguilar D, Peteiro C, Toribio J. Carcinoma de células de Merkel y linfoma no Hodgkin. Actas Dermatosifiliogr 2002; 93: 247-249
41. Brenner B, Sulkes A, Rakowsky E, Feinmesser M, Yukelson A, Bar-Haim E, Katz A, Idelevich E, Neuman A, Barhana M, Fenig E. Second neoplasms in patients with Merkel cell carcinoma. Cancer. 2001; 91: 1358-1362
42. Kayashima K, Ono T, Johno M, Kojo Y, Yamashita N, Matsunaga W. Spontaneous regression in Merkel cell (neuroendocrine) carcinoma of the skin. Arch Dermatol 1991; 127: 550-553
43. Brown TJ, Jackson BA, Macfarlane DF, Goldberg LH. Merkel cell carcinoma: spontaneous resolution and management of metastatic disease. Dermatol Surg 1999; 25: 23-25
44. Yiengpruksawan A, Coit DG, Thaler HT, et al: Merkel cell carcinoma: Prognosis and management. Arch Surg 126:1514-1519, 1991
45. Hitchcock CL, Bland KI, Laney RG III, et al: Neuroendocrine (Merkel cell) carcinoma of the skin: Its natural history, diagnosis, and treatment. Ann Surg 207:201-207, 1998
46. Alonso A, Dauden E, Álvarez-Ruiz S, Ríos L, Fraga J, García-Díez A. Placa eritematosa frontal de crecimiento rápido. Actas Dermosifiliogr 2005; 96: 264-266
47. Tyring SK, Lee PC, Omura EF, Green Lk, Merot Y. Recurrent and metastatic cutaneous neuroendocrine (Merkel cell) carcinoma mimicking angiosarcoma. Arch Dermatol. 1987; 123: 1368-1370
48. Rice RD Jr, Chonkich GD, Thompson KS, et al. Merkel cell tumor of the head and the neck. Five new cases with literature rewiew. Arch Otolaryngol Head Neck Surg. 1993; 119; 782-786
49. Ruiz R, Blasco J, Merino J, Linares J, Naranjo R. Carcinoma de células de Merkel. Presentación de seis casos. Actas Dermosifiliogr 2003; 94: 300-304
50. Balaton AJ, Capron F, Baviera EE, Meyrignac P, Vaury P, Vuong PN. Neuroendocrine carcinoma (Merkel cell tumor?) presenting as a subcutaneous tumor. An ultrastructural and immunohistochemical study of three cases. Pathol Res Pract 1989; 184: 211-216
51. Ratner D, Nelson BR, Brown MD, Johnson TM. Merkel cell carcinoma. J Am Acad Dermatol 1993; 29: 143-56
52. Poulsen M. Merkel-cell carcinoma of the skin. Lancet Oncol 2004; 5: 593-599
53. Djilali-Bouzina F, Cribier B, Heid E. Regressive neuroendocirne carcinoma after partial biopsy. Nouv Dermatol 1992; 11: 767-770
54. Duncan WC, Ttschen JA. Spontaneous regression of Merkel cell (neuroendocrine) carcinoma of the skin. J Am Acad Dermatol 1993; 29: 653–654
55. Connelly TJ, Cribier B, Brown TJ, Yanguas I. Complete spontaneous regression of Merkel cell carcinoma: A review of the 10 reported cases. Dermatol Surg 2000; 26: 853-856
56. Maruo K, Kayashima KI, Ono T. Regressing Merkel cell carcinoma-a case showing replacement of tumour cells by foamy cells. Br J Dermatol 2000; 142: 1184-1189
57. Junquera L, Torre A, Vicente JC, García-Consuegra L, Fresno MF. Complete spontaneous regression of Merkel cell carcinoma. Ann Otol Rhinol Laryngol 2005; 114: 376-380
58. O'Rourque MG, Bell JR. Merkel cell tumor with spontaneous regression. J Dermatol Surg Oncol 1986; 12: 994-996
59. Bayrou O, Avril MF, Charpentier P, Caillou B, Guillaume JC, Prade M. Primary neuroendocrine carcinoma of the skin: clinico-pathologic study of 18 cases. J Am Acad Dermatol 1991; 24: 198-207
60. Connelly TJ, Kowalcyk AP. Another case of spontaneous regression of Merkel cell (neuroendocrine) carcinoma. Dermatol Surg. 1997; 23: 588-590
61. Yanguas I, Goday JJ, Gonzalez-Guemes M, Oleaga JM, Lozano M, Soloeta R. Spontaneous regression of Merkel cell carcinoma of the skin. Br J Dermatol 1997; 137: 296-298
62. Eusebi V, Capella C, Cossu A, Rosai J: Neuroendocrine carcinoma within lymph nodes in the absence of a primary tumor, with special reference to Merkel cell carcinoma. Am J Surg Pathol 1992, 16: 658-666.
63. Díaz Iglesias JM, Fresno Forcelledo MF, Herrero-Zapatero A, Losa García JL, Díaz Iglesias C, Ablanedo Ablanedo P, Floriano Alvarez PL: Carcinoma de células de Merkel en ganglios linfáticos sin tumor cutáneo primario conocido. Presentación de 3 casos. Cirugía Española 1995; 57: 499-503
64. Heenan P J, Cole J M, Spagnolo D V. Primary cutaneous neuroendocrine carcinoma (Merkel cell tumor) an adnexal epithelial neoplasm. Am J Dermatopathol 1990: 12: 7– 16
65. Mount SL, Taatjes DJ. Neuroendocrine carcinoma of the skin (Merkel cell carcinoma). An immunoelectron-microscopic case sutdy. Am J Dermatopathol 1994; 16: 60-65
66. Kuhajda FP, Olson JL, Mann RB: Merkel cell (small cell) carcinoma of the skin: immunohistochemical and ultrastructural demonstration of distinctive perinuclear cytokeratin aggregates and a possible association with B cell neoplasms. Histochem J 1986 May; 18(5): 239-44[Medline]
67. Bickle K, Glass F, Messina JL, Fenske NA, Siegrist K. Merkel cell carcinoma: a clinical, histopathologic and immunohistochemical review. Semin Cutan Med Surg 2004; 23: 46-53
68. Drijkoningen M, de Wolf-Peeters C, van Limbergen E, Desmet V. Merkel cell tumor of the skin: an immunohistochemical study. Hum Pathol. 1986; 17: 301-307
69. Sibley RK, Dahl D. Primary neuroendocrine (Merkel cell?) carcinoma of the skin. II. An immunocytochemical study of 21 cases. Am J Surg Pathol. 1985; 9: 109-116
70. Weiler R, Fischer-Colbrie R, Schmid KW, Feichtinger H, Bussolati G, Grimelius L, Krisch K, Kerl H, O'Connor D, Winkler H. Immunological studies on the occurrence and properties of chromogranin A and B and secretogranin II in endocrine tumors. Am J Surg Pathol 1988; 12: 877-884
71. Mount SL, Taatjes DJ. Neuroendocrine carcinoma of the skin (Merkel cell carcinoma). An immunoelectron-microscopic case sutdy. Am J Dermatopathol 1994; 16: 60-65
72. Nicholson SA, McDermott MB, Swanson PE, Wick MR. CD99 and cytokeratin-20 in small-cell and basaloid tumors of the skin. Appl Immunohistochem Mol Morphol. 2000; 8: 37- 41
73. Chan JK, Suster S, Wenig BM, Tsang WY, Chan JB, Lau AL. Cytokeratin 20 immunoreactivity distinguishes Merkel cell (primary cutaneous neuroendocrine) carcinomas and salivary gland small cell carcinomas from small cell carcinomas of various sites. Am J Surg Pathol 1997; 21: 226-234
74. Leong AS, Phillips GE, Pieterse AS, Milios J. Criteria for the diagnosis of primary endocrine carcinoma of the skin ( Merkel cell carcinoma). A histological immunochemical and ultrastructural study of 13 cases. Pathology 1986;18:393-399
75. Vortmeyer AO, Merino MJ, Boni R, Liotta LA, Cavazzana A, Zhuang Z. Genetic changes associated with primary Merkel cell carcinoma. Am J Clin Pathol 1998; 109: 565-570
76. Van Gele M, Leonard JH, Van Roy N, Van Limbergen H, Van Belle S, Cocquyt V, Salwen H, De Paepe A, Speleman F. Combined karyotyping, CGH and M-FISH. Fulltext
77. Van Gele M, van Roy N, Ronan SG, et al: Molecular análisis of 1p36 breakpoints in two Merkel cell carcinomas. Genes Chromosomes Cancer 23:67-71, 1998
78. Leonard JH, Cook AL, Nancarrow D, et al: Deletion mapping on the short arm of chromosome 1 in Merkel cell carcinoma. Cancer Detect Prev 24:620-627, 2000
79. Leonard JH, Williams G, Walters MK, Nancarrow DJ, Rabitts PH. Deletion mapping of the short arm of chromosome 3 in Merkel cell carcinoma. Genes Chromosomes Cancer 1996; 15: 102-107
80. Maris JM, Matthay KK: Molecular biology of Neuroblastoma. J Clin Oncol 17:2264-2279, 1999
81. Smedley D, Sidhar S, Birdsall S, et al: Characterization of chromosome 1 abnormalities in malignant melanomas. Genes Chromosomes Cancer 28:121-125, 2000
82. Harnett PR, Kearsley JH, Dracopoli N. Loss of allelic heterozygosity on distal chromosome 1p in merkel cell carcinoma: a marker of neural crest origins. Cancer Genet Cytogenet 1991; 54: 109-113
83. Van Gele M, Speleman F, Vandesompele J, Van Roy N, Leonard JH. Characteristic pattern of chromosomal gains and losses in Merkel cell carcinoma detected by comparative genomic hybridization. Cancer Res 1998; 58: 503-1508
84. Shaw JH, Rumball E, Merkel cell tumour: Clinical behaviour and treatment. Br J Surg 78:138-142, 1991
85. Kokoska ER, Kokoska MS, Collins BT, et al: Early aggressive treatment for Merkel cell carcinoma improves outcome. Am J Surg 174:688-693, 1997
86. Mohs FE: Chemosurgery, a microscopically controlled method of cancer excision. Arch Surg 42:279-295, 1941
87. Ames SE, Krag DN, Brady MS: Radiolocalization of the sentinel lymph node in Merkel cell carcinoma: a clinical analysis of seven cases. J Surg Oncol 1998 Apr; 67(4): 251-4[Medline]
88. Cotlar AM, Gates JO, Gibbs FA Jr: Merkel cell carcinoma: combined surgery and radiation therapy. Am Surg 1986 Mar; 52(3): 159-64[Medline]
89. Smith DF, Messina JL, Perrott R, Berman CG, Reintgen DS, Cruse CW, Glass FL, Fenske NA, DeConti RC, Trotti A 3rd. Clinical approach to neuroendocrine carcinoma of the skin (Merkel cell carcinoma). Cancer Control 2000; 7: 72-83
90. Zeitouni, NC, Cheney, RT, Delacure, MD. Lymphoscintigraphy, sentinel lymph node biopsy, and Mohs micrographic surgery in the treatment of Merkel cell carcinoma. Dermatol Surg 2000; 26: 12-18
91. Rodrigues LK, Leong SP, Kashani-Sabet M, Wong JH. Early experience with sentinel lymph node mapping for Merkel cell carcinoma. J Am Acad Dermatol 2001; 45: 303-308
92. Pfeifer T, Weinberg H, Brady MS: Lymphatic mapping for Merkel cell carcinoma. J Am Acad Dermatol 37:650-651, 1997
93. Messina JL, Reintgen DS, Cruse CW, et al: Selective lymphadenectomy in patiens with Merkel cell (cutaneous neuroendocrine) carcinoma. Ann Surg Oncol 4:389-395, 1997
94. Bilchik AJ, Giuliano A, Essner R, et al: Universal application of intraoperative lymphatic mapping and sentinel lymphadenectomy in solid neoplasms. Cancer J Sei Am 4:351-358, 1998
95. Tai PT, Yu E, Tonita J, Gilchrist J. Merkel cell carcinoma of the skin. J Cutan Med Surg. 2000; 4: 186-195
96. Westgate SJ. Radiation therapy for skin tumors. Otolaryngol Clin North Am 1993; 26: 295-309
97. Crown J, Lipzstein R, Cohen S, et al: Chemotherapy of metastatic Merkel cell cancer. Cancer Invest 1991; 9(2): 129-32[Medline]
98. Paradela S, Peña C, Fernández-Jorge B, Vieira V, Rodríguez-Lozano J, García-Rozado A, Fonseca E. Carcinoma de células de Merkel. Actas Dermosifiliogr 2004; 95: 553-559
99. Fenig E, Brenner B, Katz A, Rakovsky E, Hana MB, Sulkes A. The role of radiation therapy and chemotherapy in the treatment of Merkel cell carcinoma. Cancer 1997; 80: 881–885
100. Allen PJ, Bowne WB, Jaques DP, Brennan MF, Busam K, Coit DG. Merkel Cell Carcinoma: Prognosis and treatment of patients from a single institution. J Clin Oncol 2005; 23: 2300-2309
101. Llombart B, Monteagudo C, López-Guerrero JA, Carda C, Jorda E, Sanmartín O, Almenar S, Molina I, Martín JM, Llombart-Bosch A. Clinicopathological and immunohistochemical analysis of 20 cases of Merkel cell carcinoma in search of prognostic markers. Histopathology. 2005; 46: 622-634
102. Gould VE, Moll R, Moll I, Lee I, Franke WW. Neuroendocrine (Merkel) cells of the skin: hyperplasias, dysplasias, and neoplasms. Lab Invest 1985; 52: 334-353
103. Bahadir S, Çobanoglu Ü, Alpay K, Özoran Y, Parlat P, Aykanat D. Merkel cell carcinoma. J Eur Acad Dermatol Venerol. 2003; 17: 602-603
104. Mott RT, Smoller BR, Morgan MB. Merkel cell carcinoma: a clinicopathologic study with prognostic implications. J Cutan Pathol. 2004; 31; 217–223
105. Pilotti S, Rilke F, Bartoli C, Grisotti A. Clinicopathologic correlations of cutaneous neuroendocrine Merkel cell carcinoma. J Clin Oncol 1988; 6: 1863–1873
106. Gollard R, Weber R, Kosty MP, Greenway HT, Massullo V, Humberson C. Merkel cell carcinoma: review of 22 cases with surgical, pathologic, and therapeutic considerations. Cancer 2000; 88:1842-1851
107. Woodworth B, Lacey JP, Amedee RG. Merkel cell carcinoma: an overview and case report. J La State Med Soc 2001; 153: 522-526
108. Smith KJ, Skelton HG 3rd, Holland TT, Morgan AM, Lupton GP. Neuroendocrine (Merkel cell) carcinoma with an intraepidermal component. Am J Dermatopathol 1993; 15: 528-533
109. Ferringer T, Rogers HC, Metcalf JS. Merkel cell carcinoma in situ. J Cutan Pathol 2005; 32: 162–165
110. Sidhu GS, Feiner H, Flotte TJ, Mullins JD, Schaefler K, Schultenover SJ. Merkel cell neoplasms. Histology, electron microscopy, biology, and histogenesis. Am J Dermatopathol 1980; 2: 101-119
111. Frigerio B, Capella C, Eusebi V, Tenti P, Azzopardi JG. Merkel cell carcinoma of the skin: the structure and origin of normal Merkel cells. Histopathology 1983; 7: 229-249
112. Warner TF, Uno H, Hafez GR, Burgess J, Bolles C, Lloyd RV, Oka M. Merkel cells and Merkel cell tumors. Ultrastructure, immunocytochemistry and review of the literature. Cancer 1983; 52: 238-245
113. Kossard S, Wittal R, Killingsworth M. Merkel cell carcinoma with a desmoplastic portion. Am J Dermatopathol 1995; 17: 517-522
114. Sibley RK, Dehner LP, Rosai J. Primary neuroendocrine (Merkel cell?) carcinoma of the skin. I. A clinicopathologic and ultrastructural study of 43 cases. Am J Surg Pathol. 1985; 9: 95-108
115. Goessling W, McKee PH, Mayer RJ. Merkel cell carcinoma. J Clin Oncol 2002; 20: 588-598
116. Schmid C, Beham A, Feichtinger J, et al: Recurrent and subsequently metastasizing Merkel cell carcinoma in 7-year-old girl. Histopathology 20:437-439, 1992
117. Sonak RA, Trede K, Gerharz CD: Merkel cell tumor of the hand in a 104-year-old patient: Case report with review of the literature. Handchir Mikrochir Plast Chir 28:43-45, 1996
118. Meyer-Pannwitt U, Kummerfeldt K, Boubaris P, et al: Merkel cell tumor or neuroendocrine skin carcinoma. Langenbecks Arch Chir 382:349.358, 1997
119. Ott MJ, Tanabe KK, Gadd MA, et al: Multimodality management of Merkel cell carcinoma. Arch Surg 134:388-393, 1999
120. Boyle F, Pendlebury S, Bell: Further insights into the natural history and management of primary cutaneous neuroendocrine (Merkel cell) carcinoma. Int J Radiat Oncol Biol Phys 31:315-323, 1995
121. Kroll MH, Toker C: Trabecular carcinoma of the skin: Further clinicopathologic and morphologic study. Arch Pathol Lab Med 106:404-408, 1982
122. Gooptu C, Woollons A, Ross J, Price M, Wojnarowska F, Morris PJ, Wall S, Bunker CB. Merkel cell carcinoma arising after therapeutic immunosuppression. Br J Dermatol 1997; 137: 637-641
123. Allen PJ, Zhang ZF, Coit DG.Surgical management of Merkel cell carcinoma. Ann Surg 1999; 229:97-105. full text
124. Meeuwissen JA, Bourne RG, Kearsley JH. The importance of postoperative radiation therapy in the treatment of Merkel cell carcinoma. Int J Radiat Oncol Biomed. Am J Surg 1997; 174:688-93 abstract
125. Koljonen V, Bohling T, Granhroth G, Tukiainen E. Merkel cell carcinoma: a clinicopathological study of 34 patients. Eur J Surg Oncol 2003; 29: 607-610
126. Savage P, Constenla D, Fisher C, Thomas JM, Gore ME.The natural history and management of Merkel cell carcinoma of the skin: a review of 22 patients treated at the Royal Marsden Hospital. Clin Oncol (R Coll Radiol) 1997;9:164-7
127. Pacella J, Ashby M, Ainslie J, Minty C. The role of radiotherapy in the management of primary cutaneous neuroendocrine tumors (Merkel cell or trabecular carcinoma): experience at the Peter MacCallum Cancer Institute (Melbourne, Australia). Int J Radiat Oncol Biol Phys 1988; 14:1077-84. abstract
128. Bischof M, van Kampen M, Huber P, Wannenmacher M. Merkel cell carcinoma: the role of radiation therapy in general management. Strahlenther Onkol 1999; 175:611-5. abstract
129. Walsh NM. Primary neuroencocrine (Merkel cell) carcinoma of the skin: morphologic diversity and implications thereof. Hum Pathol 2001; 32: 680-689
130. Tai PT, Yu E, Winquist E, Hammond A, Stitt L, Tonita J, Gilchrist J. Chemotherapy in neuroendocrine/Merkel cell carcinoma of the skin: case series and review of 204 cases. J Clin Oncol 2000; 18: 2493-2499.
131. Brissett AE, Olsen KD, Kasperbauer JL, Lewis JE, Groellner JR, Spotts BE, et al. Merkel cell carcinoma of the head and neck: a retrospective series. Head Neck 2002; 24: 982-988
132. Poulsen M, Rischin D, Walpole E, Harvey J, Mackintosh J, Ainslie J, Hamilton C, Keller J, Tripcony L, Trans-Tasman Radiation Oncology Group. High-risk Merkel cell carcinoma of the skin treated with synchronous carboplatin/etoposide and radiation: a Trans-Tasman Radiation Oncology Group Study--TROG 96:07. J Clin Oncol 2003; 21: 4371-4376
133. O'Connor, WJ, Roenigk, RK, Brodland, DG. Merkel cell carcinoma. Comparison of Mohs micrographic surgery and wide excision in eighty-six patients. Dermatol Surg 1997; 23: 929-923
134. Hapcic K, Panchal J, Stewart J, Levine N. Giant Merkel cell carcinoma involving the upper extremity. Dermatol Surg 2001; 27: 493-494
135. Eng TY, Boersma MG, Fuller CD, Cavanaugh SX, Valenzuela F, Herman TS. Treatment of merkel cell carcinoma. Am J Clin Oncol 2004; 27: 510-515
136. O'Brien PC, Denham JW, Leong AS. Merkel cell carcinoma: a review of behaviour patterns and management strategies. Aust N Z J Surg 1987; 57:847-50. abstract
137. Smith PD, Patterson JW. Merkel cell carcinoma (neuroendocrine carcinoma of the skin). Am J Clin Pathol 2001; 115 Suppl: S68-78
138. Benessahraoui M, Dalstein V, Lorchel F, Algros MP. Puzenat E. Louvat P. Hassam B. Humbert PH. Aubin F. Carcinoma à cellules de Merkel: etude descriptive de 24 cas. Rev Med Interne 2003; 24: 560-568
139. Smith DE, Bielamowicz S, Kagan AR, Anderson PJ, Peddada AV. Cutaneous neuroendocrine Merkel cell carcinoma: a report of 35 cases. Am J Clin Oncol 1995; 18:199-203. abstract
140. Fernández-Figueras MT, Puig L, Musulen E, Gilaberte M, Ferrándiz C, Lerma E, Ariza A. Prognostic significance of p27Kip1, p45Skp2 and Ki67 expression profiles in Merkel cell carcinoma, extracutaneous small cell carcinoma, and cutaneous squamous cell carcinoma. Histopathology. 2005; 46, 614–621
141. Eftekhari F, Wallace S, Silva EG, Lenzi R. Merkel cell carcinoma of the skin: imaging and clinical features in 93 cases. Br J Radiol 1996; 69:226-33. abstract
142. Krasagakis K, Almond-Roesler B, Zouboulis CC, Tebbe B, Wartenberg E, Wolff KD, et al. Merkel cell carcinoma: report of ten cases with emphasis on clinical course, treatment, and in vitro drug sensitivity. J Am Acad Dermatol 1997; 36:727-32.fulltext
143. Hauschild A, Rademacher D, Rowert J, et al: Merkel cell carcinoma: Follow-up of 10 patiens-Current diagnosis and theraphy. Langenbecks Arch Chir 382:185-195
144. Bertó J, Cuenca A, Díaz-Martínez B, Peña ML, Ruíz-Fernández P, Sánchez de Paz F. Carcinoma de células de Merkel. Estudio de cinco casos. Actas Dermosifiliogr 2005; 96: 106-110
145. Morrison WH, Peters LJ, Silva EG, Wendt CD, Ang KK, Goepfert H. The essential role of radiation therapy in securing locoregional control of Merkel cell carcinoma. Int J Radiat Oncol Biol Phys 1990; 19: 583-591
146. Pitman MJ, Willians JD. Merkel cell carcinoma of the skin. Department of Otolaryngology, ENT and Allergy Associates.2005.Nov 4. Fulltext
147. Tennvall J, Biorklund A, Johansson L, Akerman M. Merkel cell carcinoma: management of primary, recurrent and metastatic disease: a clinicopathological study of 17 patients. Eur J Surg Oncol 1989; 15:1-9. abstract
Anexos:
|