Rebeca nació en Santo Domingo a padres muy
jóvenes e impacientes. Por algunos años el embarazo fue dificultado por
una conteo bajo de esperma de parte del marido, cuyo orgullo sufriría
inmensamente, ya que algo afectando sus esencias masculinas, le ofendía.
Pero, el método de la inseminación
artificial produjo el resultado anhelado y con un donante que llenaba los
requerimientos de la pareja. Rebeca nació, “la rubita” por todos,
anticipada.
Nereida, la niñera expectante, telefoneó a
sus familiares, expresando admiración incontenible y exuberante: “¡tiene
los ojos azulitos!”
Los padres mantenían una propiedad cercana
a la nuestra en Casa de Campo, viniendo a menudo a utilizar nuestra
piscina, con Rebeca, por supuesto.
En verdad que era muy hermosa y plácida…
Como bebé, dormía constantemente y su piel resistía muy poco la luz
solar, requiriendo que se la mantuviera a la sombra la mayor parte del
tiempo.
La mamá era una mujer con tendencias al
sobrepeso, el cual combatiera con visitas al gimnasio local y con la
ingestión profusa de comidas y de bebidas dietéticas.
Temiendo que la hija, también pudiese
engordar, muy pronto, Rebeca empezaría a beber refrescos de dieta ---
como hacía la mamá.
A los dos años de edad, la niña aún no
poseía ningún signo de desarrollo del lenguaje ni caminaba sin ayuda. Su
coordinación era asimismo muy pobre.
Vinieron a vernos cuando un día se le
infectó un oído y, siendo domingo, no tenían donde llevarla. La examiné
y le prescribí antibióticos para una otitis media.
La madre, sollozando, me pidió que
examinara la hija, ya que creía que algo estaba muy mal con ella;
arrepintiéndose del embarazo artificial.
Así lo hice. El examen neurológico era
preocupante. El examen psicológico directo era ominoso.
Me confesó que el marido se había alejado
de ella y de la hija y que había expresado el deseo de una separación
marital. Eso la ponía muy triste y no sabía qué hacer para atraerlo de
nuevo.

El
diagnóstico: La fenilcetonuria
La fenilcetonuria es una enfermedad
hereditaria poco común, en la cual el cuerpo no metaboliza adecuadamente
el aminoácido fenilalanina, lo cual puede
causar retardo
mental severo.
Esta condición se hereda como un rasgo
autosómico recesivo que se encuentra en uno de cada 16,000 partos en los
Estados Unidos, con una distribución igual en ambos sexos. Dado que esta
enfermedad se puede detectar fácilmente a través de un examen de sangre
y se puede tratar, en muchas partes se exige un examen urinario para todos
los recién nacidos.
La anomalía genéticamente determinada en
la fenilcetonuria es la ausencia de la enzima
denominada fenilalanina
hidroxilasa. La fenilalanina es uno de los ocho aminoácidos esenciales
encontrados en los alimentos que contienen proteínas. En este trastorno,
la fenilalanina no se puede metabolizar en forma normal, debido a la
ausencia de esta enzima. Por consecuencia, los niveles de fenilalanina se
elevan y se desarrollan dos substancias derivadas estrechamente
relacionadas con la fenilalanina. Estas sustancias son tóxicas para el sistema nervioso central y
pueden ocasionar daño cerebral.
El daño cerebral es causante de una
variedad de retardo mental severo que
se detecta hacia el final del primer año de vida. Los niños mayores
pueden desarrollar trastornos motores, de la coordinación e hiperactividad. Debido a
que la fenilalanina está comprometida de forma indirecta en la producción
de la melanina, el pigmento
responsable del color de la piel y el cabello, los niños con
fenilcetonuria tienen una complexión más clara (cabello rubio y ojos
azules) que la de los hermanos no afectados. También pueden emitir un
olor muy desagradable que resulta de la acumulación de ácido fenilacético
y que se puede sentir en el aliento, la piel y la orina.

Cabellos
rubios, ojos azules, lesiones de la piel.
Prevención
Se
recomienda la asesoría genética para los futuros padres con antecedentes
familiares de fenilcetonuria. El estado portador se puede detectar por
medio de pruebas enzimáticas y además, se
puede diagnosticar en forma prenatal. En los Estrados Unidos, se deben
realizar rutinariamente las pruebas de tamizaje a todos los bebés
inmediatamente después del nacimiento.
Es
necesario que todas las madres gestantes portadoras de
fenilcetonuria sigan estrictamente una dieta especial baja en fenilalanina ya que la
acumulación de este compuesto afectará al neonato, aún sin que haya
heredado la enfermedad.
Síntomas
·
erupción cutánea eczematosa
·
microcefalia
·
movimientos faltos de coordinación
de brazos y piernas
·
tono muscular espástico
·
postura inusual de las manos
·
retardo del desarrollo intelectual
y social
·
retardo en el desarrollo del habla
y del lenguaje
·
un fuerte olor desagradable en la
orina y el sudor
·
coloración pálida.
Signos
diagnósticos
La apariencia es diagnóstica, donde la
enfermedad se sospecha: complexión pálida, cabello rubio y ojos azules.
Los
pruebas de laboratorios indicadas
·
análisis enzimático para
detectar el estado de portador
·
prueba de las vellosidades coriónicas
del embarazo materno, para detectar fenilcetonuria en el feto
·
tamizaje para fenilcetonuria. Esta
consiste en la obtención de una muestra de sangre del talón del bebé,
extraída con una lanceta, para tamizaje, obligatorio en la mayoría de
los estados de los Estados Unidos
Tratamiento
El
tratamiento comprende una dieta libre de fenilalanina, especialmente
durante el crecimiento del niño para reducir o prevenir el riesgo del retardo
mental. Es necesario llevar estrictamente la dieta, por lo cual
se requiere la supervisión muy de cerca por parte del médico o el
dietista con la cooperación de los padres del niño.
La
fenilalanina se encuentra en cantidades significativas en alimentos como
la leche, los huevos y algunos comestibles comunes. Además se encuentra
en el edulcorante Nutrasweet (aspartame), razón por la cual los productos que
contengan aspartame se deben evitar en las dietas de los niños con esta
enfermedad.
Cuando
le ofreciera refrescos a su hija, la mamá no lo sabía.
Lofenalac
es una fórmula infantil especial para niños con fenilcetonuria. Se puede
usar durante toda la vida como fuente de proteína
con un contenido extremadamente bajo en fenilalanina y balanceado para los
aminoácidos esenciales
restantes.
Las
mujeres adultas portadoras de fenilcetonuria que tienen planes de salir embarazadas deben seguir
una dieta rigurosa baja en fenilalanina que debe iniciarse antes del
embarazo y continuarse durante todo el embarazo.
Pronóstico
El
futuro puede ser optimista, si el tratamiento dietético se inicia
inmediatamente después del nacimiento del bebé y se sigue estrictamente.
Pero si se comienza después de los 3 años o no se hace ningún
tratamiento, el daño cerebral es inevitable.
Complicaciones
Sin
tratamiento, el niño sufrirá retardo mental severo.
Situaciones
que requieren asistencia médica
Se
debe buscar asistencia médica si a un bebé no le han realizado exámenes
para fenilcetonuria, especialmente si algún familiar padece esta
enfermedad.
El
Caso de Aníbal, el Hiperactivo Atolondrado
“Cuando nadie
me cree, mi doctor me creerá”… El lamento de Aníbal.
En los años
1920s, en los Estados Unidos se inició un proyecto, asociado con las
cortes para juveniles que se conocería por muchos años como El
Movimiento de Orientación Infantil (The Child Guidance Movement).
Este programa
consistió de construir bajo la supervisión de las cortes, un sistema de
clínicas para asesoramiento y tratamiento del niño a riesgo y sus
familiares.
La estructura de
las clínicas era multidisciplinaria, dotadas con la presencia de un
equipo de profesionales que incluía: 1. Psiquiatra 2. Psicólogo 3.
Trabajador social, y 4. Patólogo del lenguaje.
Debido, a que la
psiquiatría de niños haría su presencia formal a fines de los años
1950s y de que existiera una demanda para el entrenamiento de
profesionales capacitados dentro de esa especialidad. Muchas universidades
norteamericanas pronto abrirían sus propias clínicas, asumirían el
control de las ya existentes o iniciarían unidades especializadas para
atraer a los candidatos futuros.
Yo me entrené
en la Clínica de la famosa Washington
University Medical School en
St. Louis. Teniendo la posterior fortuna de haber dirigido cinco centros
de tratamientos psiquiátricos para adolescentes y niños en el estado de
Missouri.
El
proceso de evaluación y tratamiento
El mecanismo de
la evaluación seguía una rutina afianzada formalmente por la tradición:
-
Llamada de referir el paciente
-
El caso se pone en la lista de espera
-
La entrevista, en grupo, de pacientes nuevos
-
La admisión como paciente
-
Otra espera
-
La evaluación
-
La conferencia diagnóstica
-
La conferencia post-diagnóstica, donde se imparten los hallazgos y las recomendaciones de tratamiento a los padres
-
La espera
-
La asignación del caso a: Un psiquiatra, el niño, y trabajador social a los padres.
-
En caso de necesitar terapia del lenguaje, por falta de personal y de fondos, el niño se refería a una de las tantas clínicas que existían en la comunidad.
Aníbal se
refirió a la clínica por su maestra. Ella había notado, que a la edad
de seis años, y luego del nacimiento de una hermana menor (de tres años,
cuando se iniciara el proceso) el habla, la socialización, la coordinación
y el temperamento de Aníbal habían sufrido deterioro aparente y
progresivo.
El proceso
sistemático de la evaluación procedería de modo ritualizado, sin poder
ser “contaminado” por contactos interdisciplinarios prior a la
conferencia diagnóstica. Toda la información proveniente del examen del
niño (de cualquier edad) estaría restringida al residente de psiquiatría,
la de los padres estaría limitada a la trabajadora social, en este caso.
Cuando fui a
encontrar a mi paciente, por la vez primera, reconocí a un niño de
cabeza muy grande, de excitabilidad fácil y con muy poca coordinación.
El camino a mi
oficina requería que bajáramos un tramo de escalones para lo que tuve
que cargarlo en mis brazos.
Entrando a la
oficina, luciría desorientado y como si estuviera perdido. Le ofrecí un
juguete para iniciar contacto social. Le di un teléfono, que adquiriría
significado especial.

Aníbal parecía
ensimismado con el sonido del timbre que su teléfono produjera cuando
discaba un número. Lo hacía repetida e incesantemente, poniendo el teléfono
al oído, como si para escuchar mejor.
Cuando lo
conduje a la casa de muñecas, que en la oficina tuviera, para
introducirlo a la familia de marionetas, me di cuenta de que no podía ver
bien.

Decidí proceder
entonces, de una manera poco convencional y llamé a la mamá que esperaba
en el segundo piso.
Ella vino y me
asistió con su hijo.
La
circunferencia cefálica estaba hipertrófica,

Cabeza
hipertrófica a la derecha
En la piel tenía
varias marcas conocidas como café
au lait spots comunes en la neurofribromatosis,
Los nervios ópticos
estaban atrofiados bilateralmente, en el examen fondoscópico del ojo.
Diagnóstico:
Glioma del nervio óptico
Confirmación
radiográfica
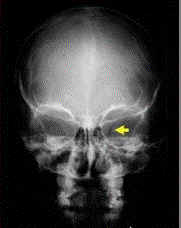
El
profesor Juan Taveras, el Neurorradiólogo – de origen dominicano -- más
destacado del mundo, confirmó el diagnóstico
Glioma
del Nervio Óptico… ¿Qué es?
Crecimiento
progresivo de una masa tumoral que invade y presiona el nervio óptico y
las estructuras adyacentes.
Síntomas
Los
síntomas son debidos a la presión que ocasiona el crecimiento tumoral:
-
Pérdida de visión en uno o ambos ojos
-
Estrabismo
-
Protuberancia ocular
Los exámenes físicos
demuestran el déficit neurológico y la atrofia típica del nervio óptico.
Signos de la
neurofribromatosis, como son las manchas de café
au lait, son frecuentes.
Diagnóstico:
TC y IRM, con
las radiografías son los métodos más confiables para llegar a un diagnóstico
acertado. La formación conocida como la “Silla Turca en J” es
patognomónica.
Tratamiento
La radioterapia
y la cirugía son los métodos de tratamientos de uso más frecuentes.
El pronóstico
es serio, como lo fuera en este caso.
Resumen
del caso de Aníbal
El teléfono
rojo fue la vía de comunicación que Aníbal y yo emplearíamos por el
tiempo que le quedaría de vida.
Todas las tarde,
y antes de salir para mi casa, a ver a mi esposa y a mis hijos, entonces
muy jóvenes, pasaba por la habitación de mi paciente a visitarlo a él y
a conversar con sus padres y con su hermanita.
Aníbal presentía
mi llegada, comenzando a “llamarme” por teléfono ruidosamente antes
de que yo entrara a su cuarto --- para mi uso en la terapia, los padres
habían adquirido un teléfono azul.
Nosotros habábamos
por un tiempo largo. Aníbal me decía del dolor que sentía y de las náuseas
que sufriera después de sus tratamientos.
Una tarde me
preguntó:
“¿Voy a
morir?”
Le dije que sí,
que nos pasará, algún día a todos.
“No, no quiero
saber lo que pasa a todos… lo que quiero es saber si voy a morir muy
pronto…”
Con lágrimas, y
sin poder disimular mi tristeza, le dije que sí…
Me
dijo “adiós”
La siguiente mañana,
el día era fría, nevaba, y su habitación ya estaba vacía…

Así son los
casos cuando son difíciles…
Bibliografía
Suministrada por solicitud.